

Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus
- PMID: 31932676
- PMCID: PMC6965005
- DOI: 10.1038/s41477-019-0577-7
Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus
Abstract
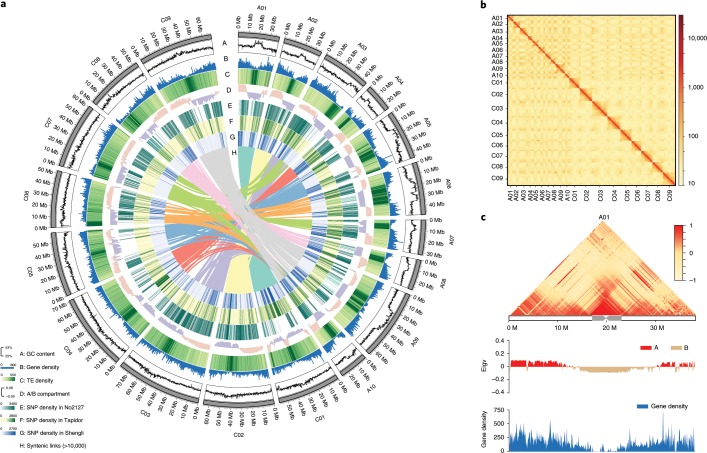
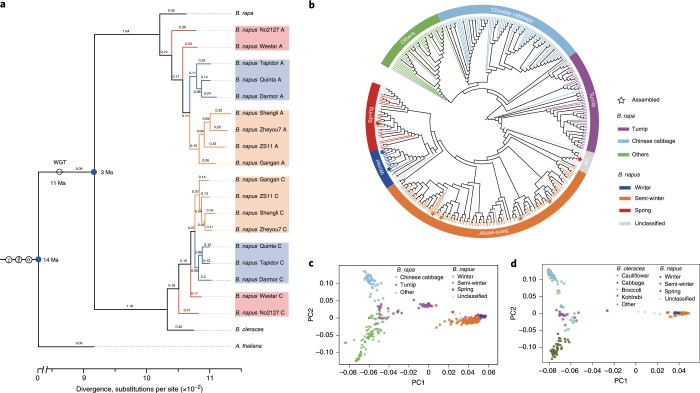
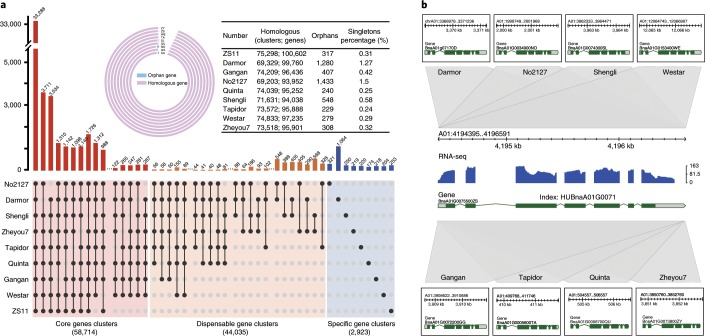
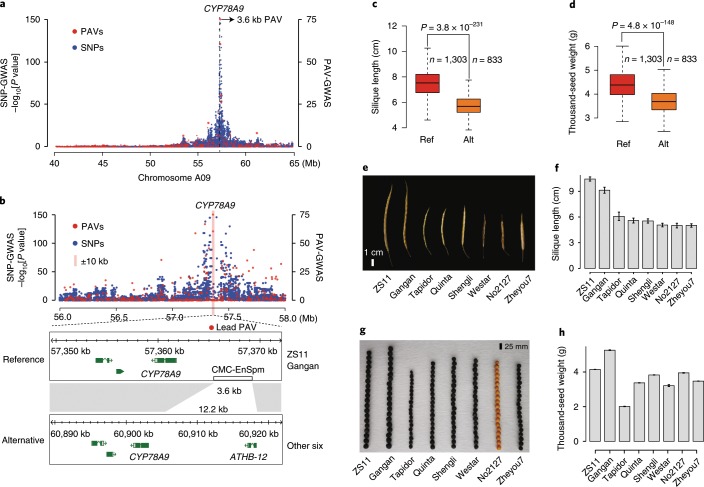
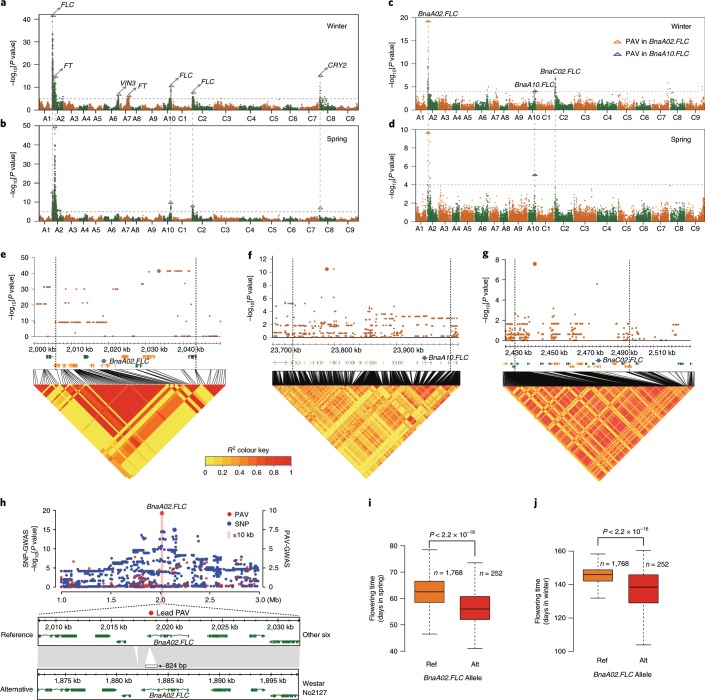
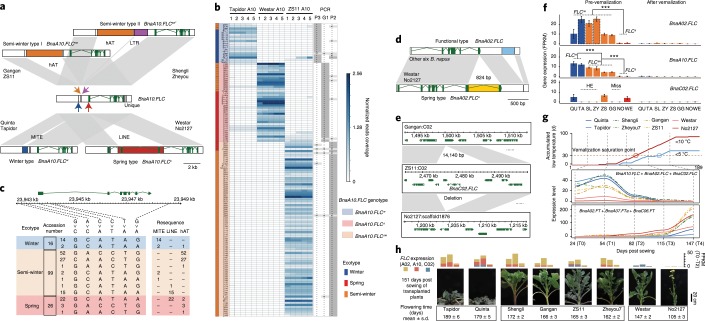
Rapeseed (Brassica napus) is the second most important oilseed crop in the world but the genetic diversity underlying its massive phenotypic variations remains largely unexplored. Here, we report the sequencing, de novo assembly and annotation of eight B. napus accessions. Using pan-genome comparative analysis, millions of small variations and 77.2-149.6 megabase presence and absence variations (PAVs) were identified. More than 9.4% of the genes contained large-effect mutations or structural variations. PAV-based genome-wide association study (PAV-GWAS) directly identified causal structural variations for silique length, seed weight and flowering time in a nested association mapping population with ZS11 (reference line) as the donor, which were not detected by single-nucleotide polymorphisms-based GWAS (SNP-GWAS), demonstrating that PAV-GWAS was complementary to SNP-GWAS in identifying associations to traits. Further analysis showed that PAVs in three FLOWERING LOCUS C genes were closely related to flowering time and ecotype differentiation. This study provides resources to support a better understanding of the genome architecture and acceleration of the genetic improvement of B. napus.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
