

Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance
- PMID: 31932713
- PMCID: PMC7047537
- DOI: 10.1038/s41564-019-0637-9
Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance
Erratum in
-
Author Correction: Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance.Nat Microbiol. 2020 Mar;5(3):525. doi: 10.1038/s41564-020-0671-7. Nat Microbiol. 2020. PMID: 31965087 Free PMC article.
Abstract
Metagenomic next-generation sequencing (mNGS), the shotgun sequencing of RNA and DNA from clinical samples, has proved useful for broad-spectrum pathogen detection and the genomic surveillance of viral outbreaks. An additional target enrichment step is generally needed for high-sensitivity pathogen identification in low-titre infections, yet available methods using PCR or capture probes can be limited by high cost, narrow scope of detection, lengthy protocols and/or cross-contamination. Here, we developed metagenomic sequencing with spiked primer enrichment (MSSPE), a method for enriching targeted RNA viral sequences while simultaneously retaining metagenomic sensitivity for other pathogens. We evaluated MSSPE for 14 different viruses, yielding a median tenfold enrichment and mean 47% (±16%) increase in the breadth of genome coverage over mNGS alone. Virus detection using MSSPE arboviral or haemorrhagic fever viral panels was comparable in sensitivity to specific PCR, demonstrating 95% accuracy for the detection of Zika, Ebola, dengue, chikungunya and yellow fever viruses in plasma samples from infected patients. Notably, sequences from re-emerging and/or co-infecting viruses that have not been specifically targeted a priori, including Powassan and Usutu, were successfully enriched using MSSPE. MSSPE is simple, low cost, fast and deployable on either benchtop or portable nanopore sequencers, making this method directly applicable for diagnostic laboratory and field use.
Conflict of interest statement
C.Y.C. is the director of the UCSF–Abbott Viral Diagnostics and Discovery Center and receives research support funding from Abbott Laboratories, Inc. X.D. and C.Y.C. are inventors on a patent application titled ‘Spiked Primer Design for Targeted Enrichment of Metagenomic Libraries’ (US application no. 62/667,344, filed 4 May 2018 by the University of California San Francisco) that includes a description of the methods and primer sets presented in this paper. A.A.A. is an employee of Karius, Inc.
Figures
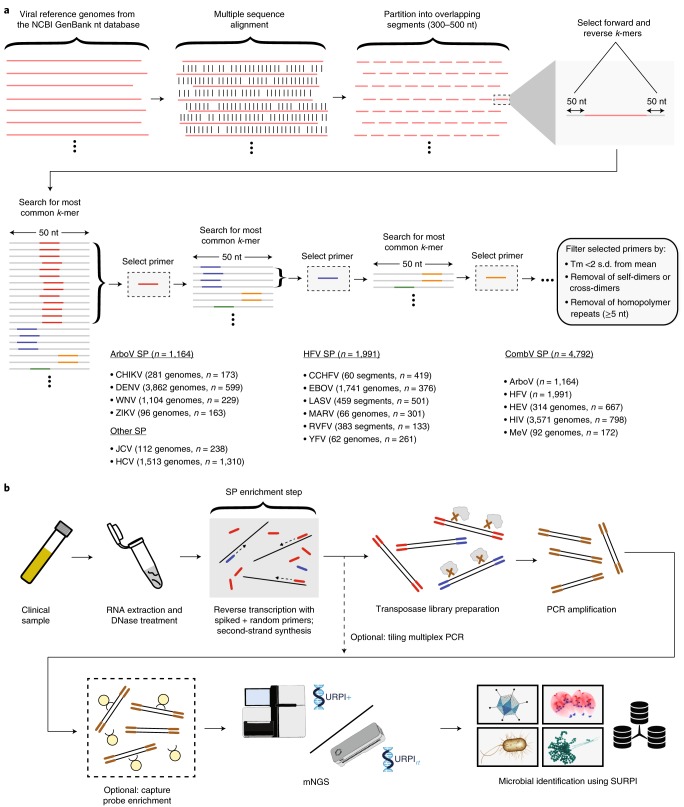

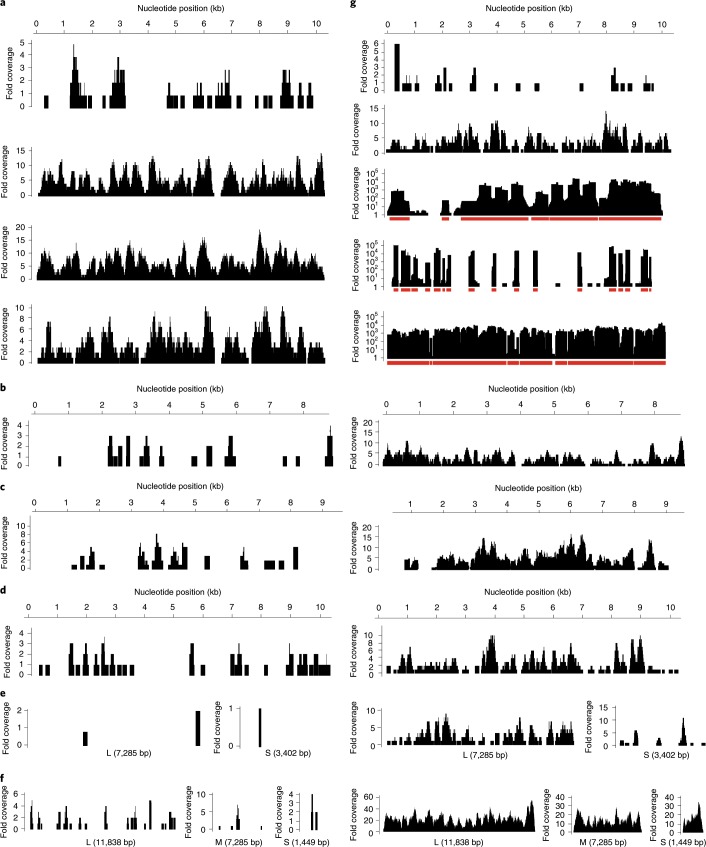
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
