

Fructose and hepatic insulin resistance
- PMID: 31935149
- PMCID: PMC7774304
- DOI: 10.1080/10408363.2019.1711360
Fructose and hepatic insulin resistance
Abstract
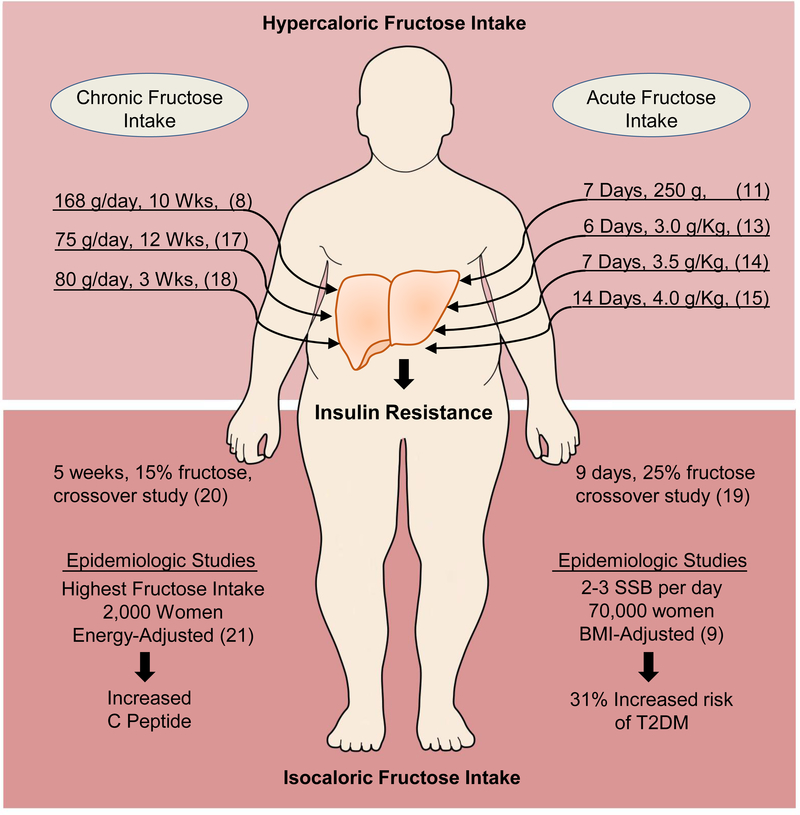
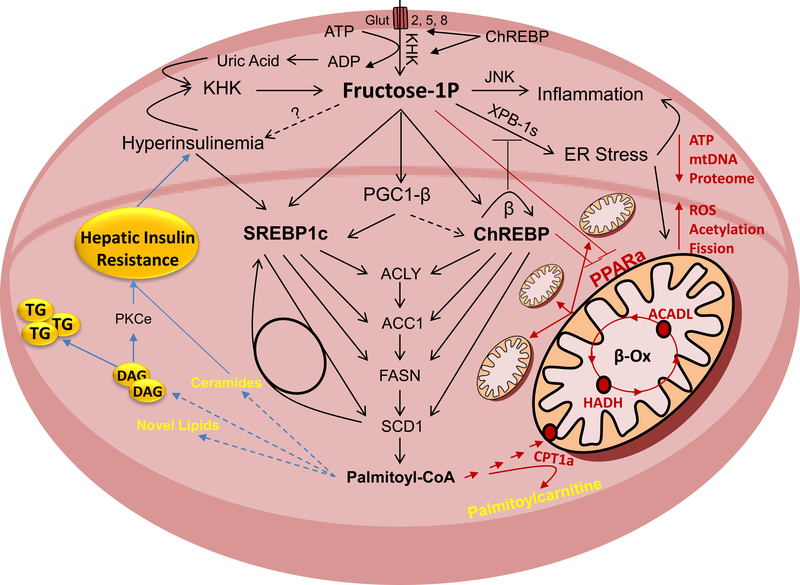
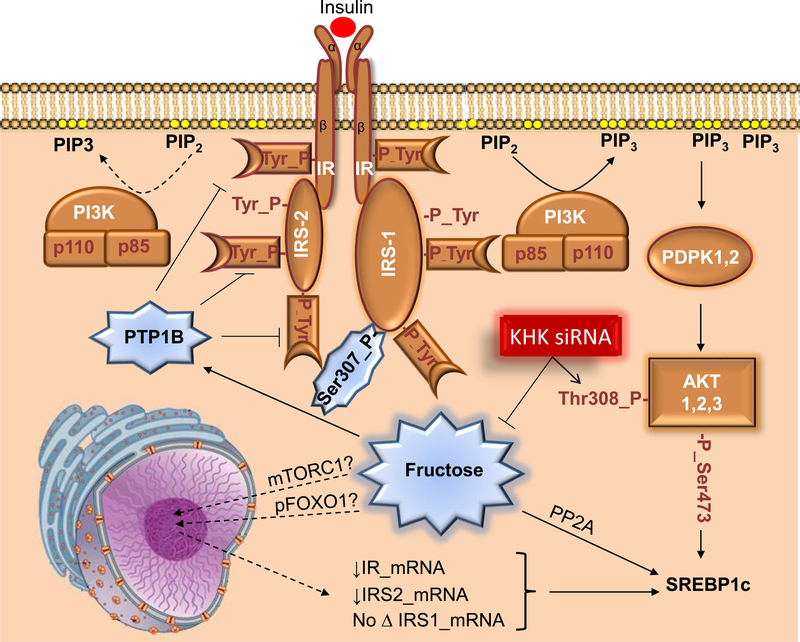
Excessive caloric intake in a form of high-fat diet (HFD) was long thought to be the major risk factor for development of obesity and its complications, such as fatty liver disease and insulin resistance. Recently, there has been a paradigm shift and more attention is attributed to the effects of sugar-sweetened beverages (SSBs) as one of the culprits of the obesity epidemic. In this review, we present the data invoking fructose intake with development of hepatic insulin resistance in human studies and discuss the pathways by which fructose impairs hepatic insulin action in experimental animal models. First, we described well-characterized pathways by which fructose metabolism indirectly leads to hepatic insulin resistance. These include unequivocal effects of fructose to promote de novo lipogenesis (DNL), impair fatty acid oxidation (FAO), induce endoplasmic reticulum (ER) stress and trigger hepatic inflammation. Additionally, we entertained the hypothesis that fructose can directly impede insulin signaling in the liver. This appears to be mediated by reduced insulin receptor and insulin receptor substrate 2 (IRS2) expression, increased protein-tyrosine phosphatase 1B (PTP1b) activity, whereas knockdown of ketohexokinase (KHK), the rate-limiting enzyme of fructose metabolism, increased insulin sensitivity. In summary, dietary fructose intake strongly promotes hepatic insulin resistance via complex interplay of several metabolic pathways, at least some of which are independent of increased weight gain and caloric intake. The current evidence shows that the fructose, but not glucose, component of dietary sugar drives metabolic complications and contradicts the notion that fructose is merely a source of palatable calories that leads to increased weight gain and insulin resistance.
Keywords: NAFLD; Sugar; fructose; insulin resistance; obesity; sugar-sweetened beverages.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous