

A practical view of fine-mapping and gene prioritization in the post-genome-wide association era
- PMID: 31937202
- PMCID: PMC7014684
- DOI: 10.1098/rsob.190221
A practical view of fine-mapping and gene prioritization in the post-genome-wide association era
Abstract
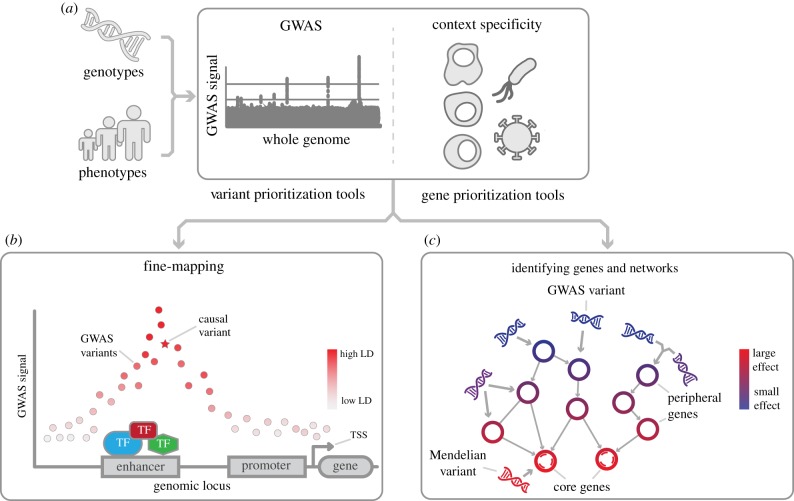
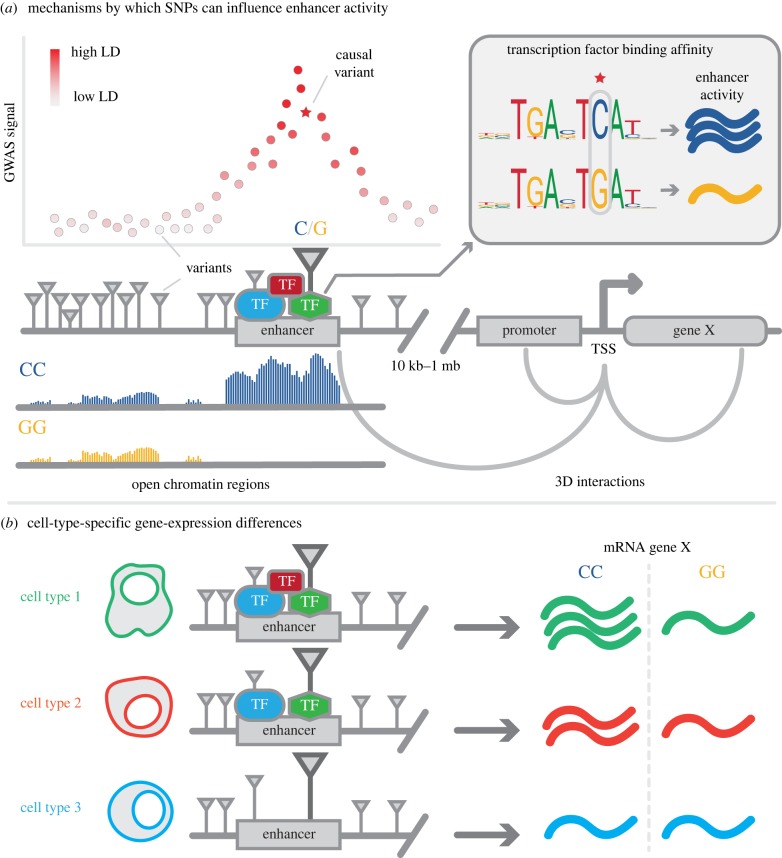
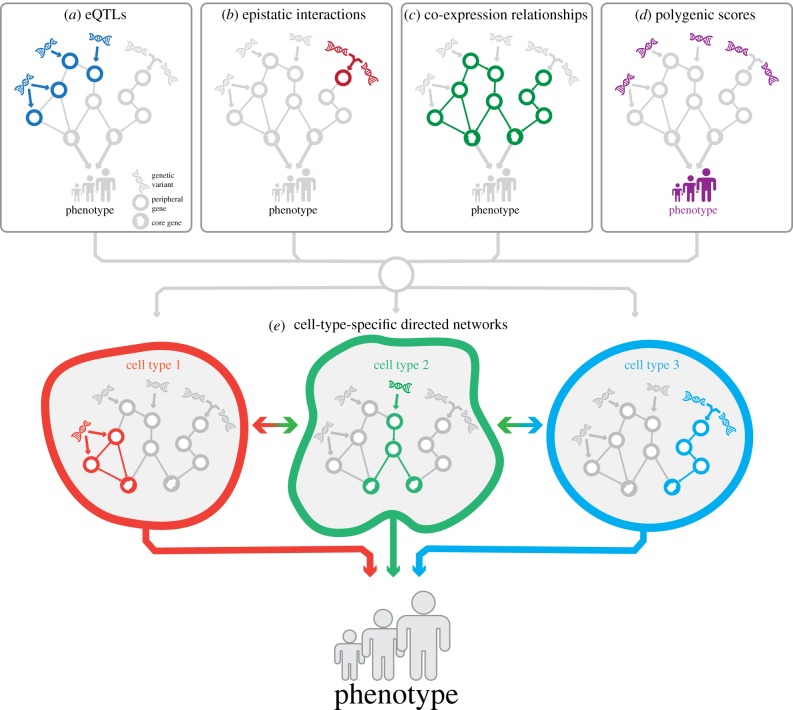
Over the past 15 years, genome-wide association studies (GWASs) have enabled the systematic identification of genetic loci associated with traits and diseases. However, due to resolution issues and methodological limitations, the true causal variants and genes associated with traits remain difficult to identify. In this post-GWAS era, many biological and computational fine-mapping approaches now aim to solve these issues. Here, we review fine-mapping and gene prioritization approaches that, when combined, will improve the understanding of the underlying mechanisms of complex traits and diseases. Fine-mapping of genetic variants has become increasingly sophisticated: initially, variants were simply overlapped with functional elements, but now the impact of variants on regulatory activity and direct variant-gene 3D interactions can be identified. Moreover, gene manipulation by CRISPR/Cas9, the identification of expression quantitative trait loci and the use of co-expression networks have all increased our understanding of the genes and pathways affected by GWAS loci. However, despite this progress, limitations including the lack of cell-type- and disease-specific data and the ever-increasing complexity of polygenic models of traits pose serious challenges. Indeed, the combination of fine-mapping and gene prioritization by statistical, functional and population-based strategies will be necessary to truly understand how GWAS loci contribute to complex traits and diseases.
Keywords: causal variants and genes; complex traits; fine-mapping; genome-wide association study; polygenic diseases; single-nucleotide polymorphisms.
Conflict of interest statement
We declare we have no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous