

Quantitative Framework for Model Evaluation in Microbiology Research Using Pseudomonas aeruginosa and Cystic Fibrosis Infection as a Test Case
- PMID: 31937646
- PMCID: PMC6960289
- DOI: 10.1128/mBio.03042-19
Quantitative Framework for Model Evaluation in Microbiology Research Using Pseudomonas aeruginosa and Cystic Fibrosis Infection as a Test Case
Abstract
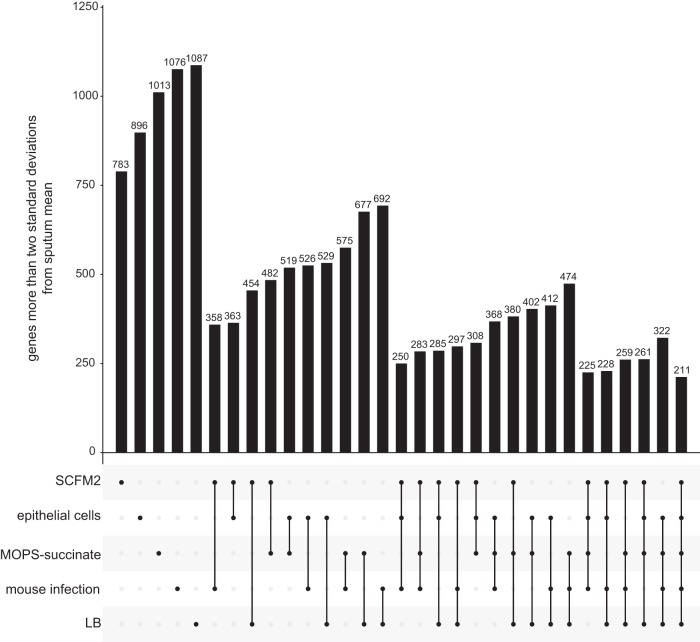
Laboratory models are a cornerstone of modern microbiology, but the accuracy of these models has not been systematically evaluated. As a result, researchers often choose models based on intuition or incomplete data. We propose a general quantitative framework to assess model accuracy from RNA sequencing data and use this framework to evaluate models of Pseudomonas aeruginosa cystic fibrosis (CF) lung infection. We found that an in vitro synthetic CF sputum medium model and a CF airway epithelial cell model had the highest genome-wide accuracy but underperformed on distinct functional categories, including porins and polyamine biosynthesis for the synthetic sputum medium and protein synthesis for the epithelial cell model. We identified 211 "elusive" genes that were not mimicked in a reference strain grown in any laboratory model but found that many were captured by using a clinical isolate. These methods provide researchers with an evidence-based foundation to select and improve laboratory models.IMPORTANCE Laboratory models have become a cornerstone of modern microbiology. However, the accuracy of even the most commonly used models has never been evaluated. Here, we propose a quantitative framework based on gene expression data to evaluate model performance and apply it to models of Pseudomonas aeruginosa cystic fibrosis lung infection. We discovered that these models captured different aspects of P. aeruginosa infection physiology, and we identify which functional categories are and are not captured by each model. These methods will provide researchers with a solid basis to choose among laboratory models depending on the scientific question of interest and will help improve existing experimental models.
Keywords: Pseudomonas aeruginosa; cystic fibrosis; infection; model; transcriptomics.
Copyright © 2020 Cornforth et al.
Figures
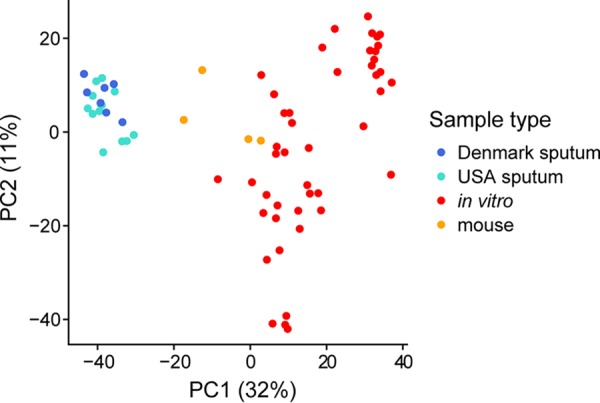
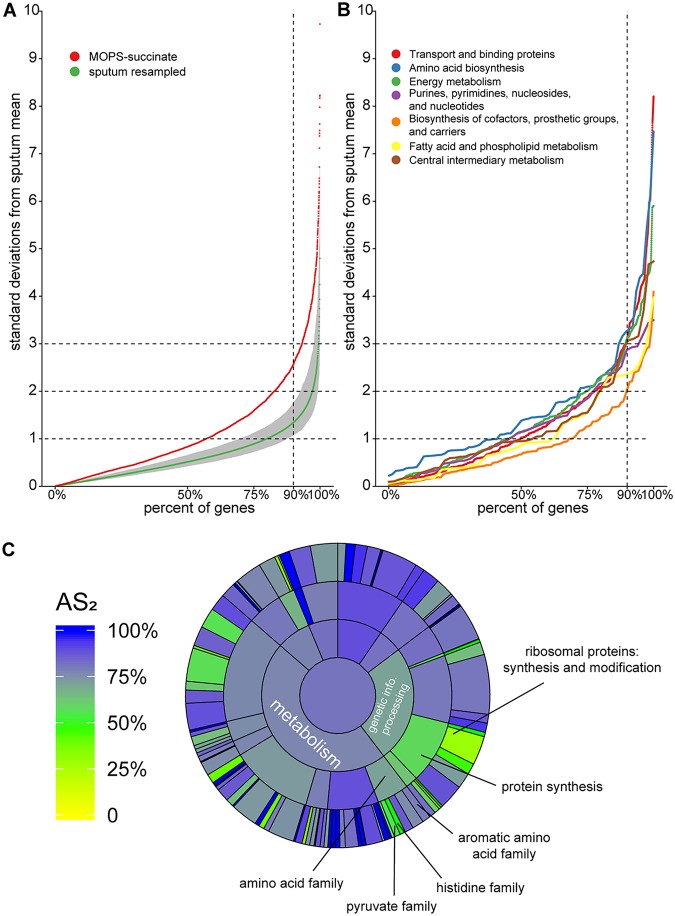
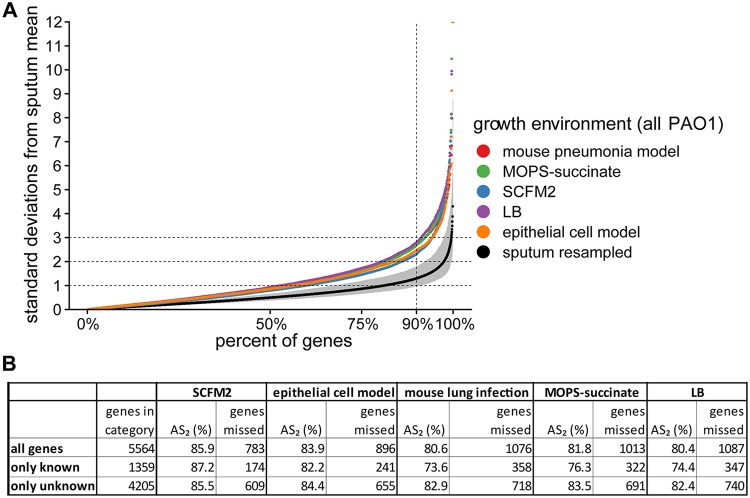
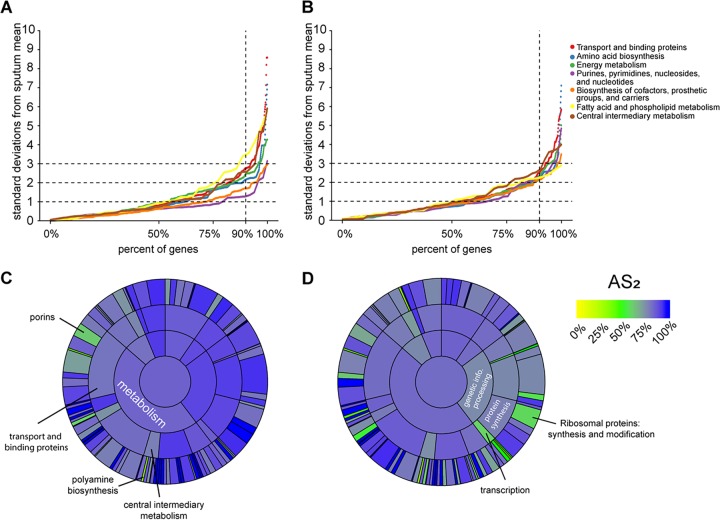
References
-
- Zak O, Sande MA. 1999. Handbook of animal models of infection. Elsevier, New York, NY.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
