

Quantitative PCR provides a simple and accessible method for quantitative microbiota profiling
- PMID: 31940382
- PMCID: PMC6961887
- DOI: 10.1371/journal.pone.0227285
Quantitative PCR provides a simple and accessible method for quantitative microbiota profiling
Abstract
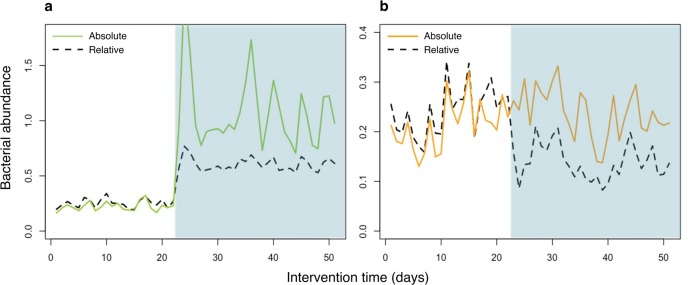
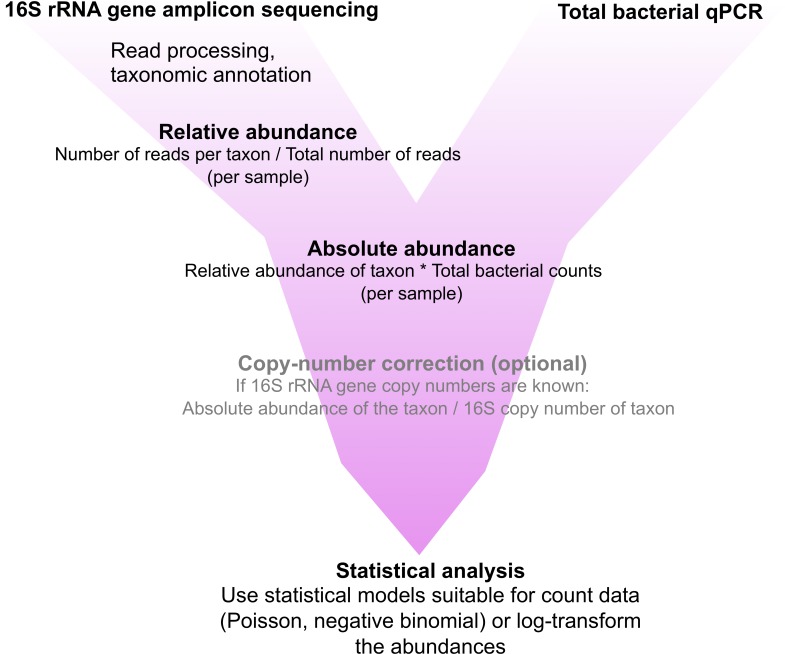
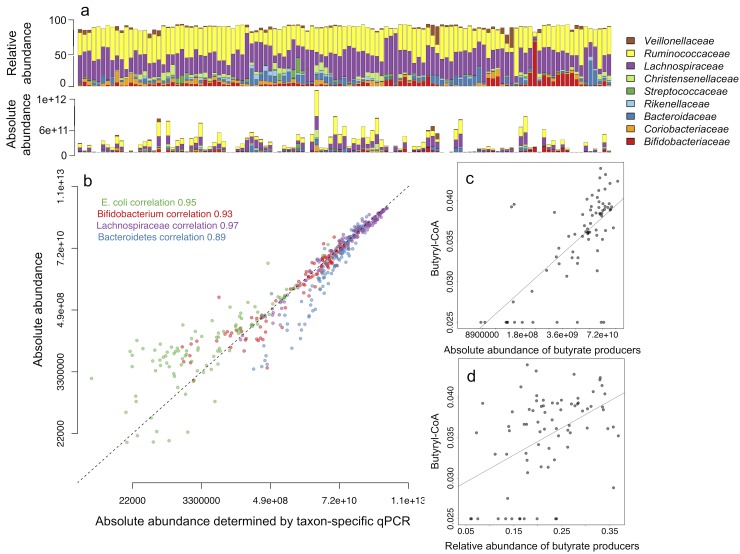
The use of relative abundance data from next generation sequencing (NGS) can lead to misinterpretations of microbial community structures, as the increase of one taxon leads to the concurrent decrease of the other(s) in compositional data. Although different DNA- and cell-based methods as well as statistical approaches have been developed to overcome the compositionality problem, and the biological relevance of absolute bacterial abundances has been demonstrated, the human microbiome research has not yet adopted these methods, likely due to feasibility issues. Here, we describe how quantitative PCR (qPCR) done in parallel to NGS library preparation provides an accurate estimation of absolute taxon abundances from NGS data and hence provides an attainable solution to compositionality in high-throughput microbiome analyses. The advantages and potential challenges of the method are also discussed.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
