

A benchmark of batch-effect correction methods for single-cell RNA sequencing data
- PMID: 31948481
- PMCID: PMC6964114
- DOI: 10.1186/s13059-019-1850-9
A benchmark of batch-effect correction methods for single-cell RNA sequencing data
Abstract
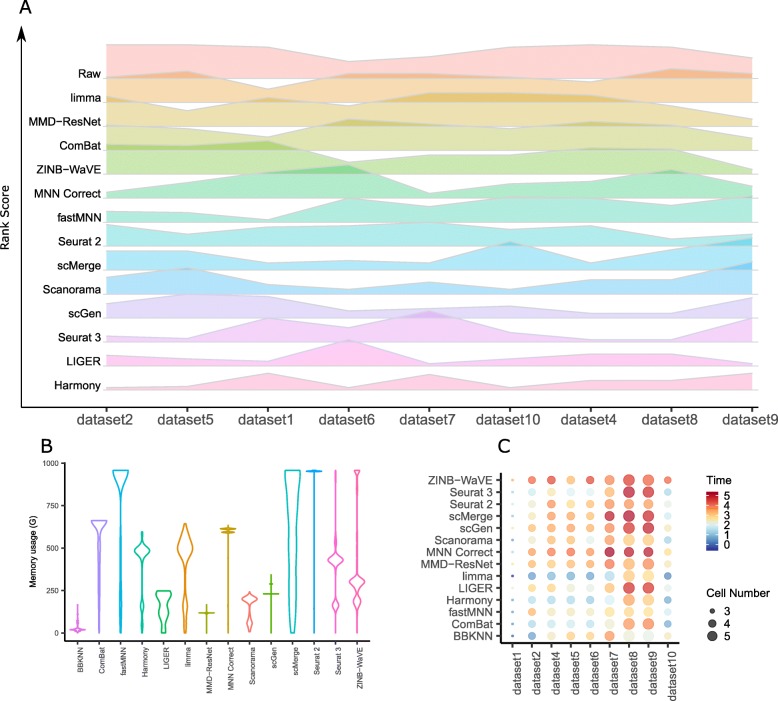
Background: Large-scale single-cell transcriptomic datasets generated using different technologies contain batch-specific systematic variations that present a challenge to batch-effect removal and data integration. With continued growth expected in scRNA-seq data, achieving effective batch integration with available computational resources is crucial. Here, we perform an in-depth benchmark study on available batch correction methods to determine the most suitable method for batch-effect removal.
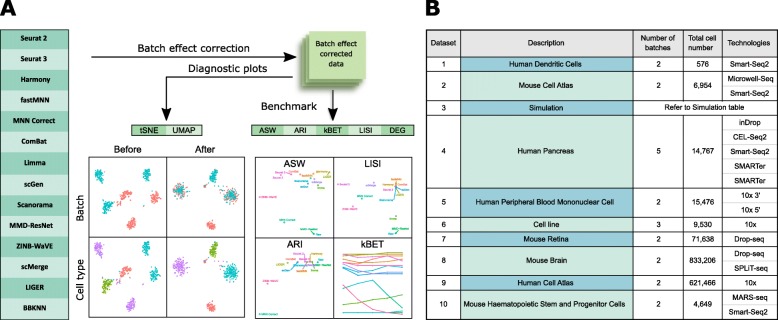
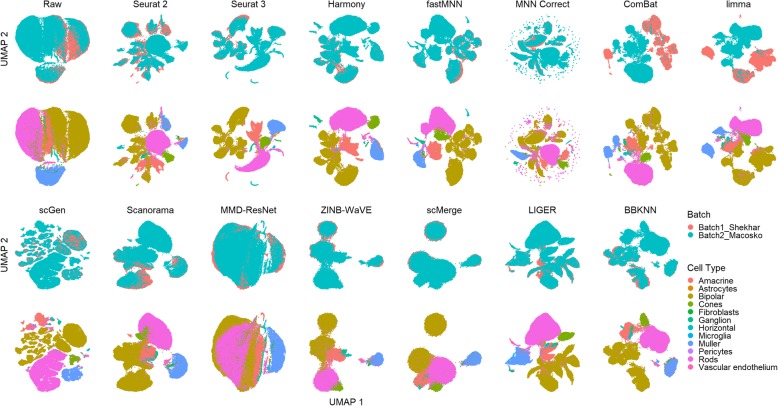
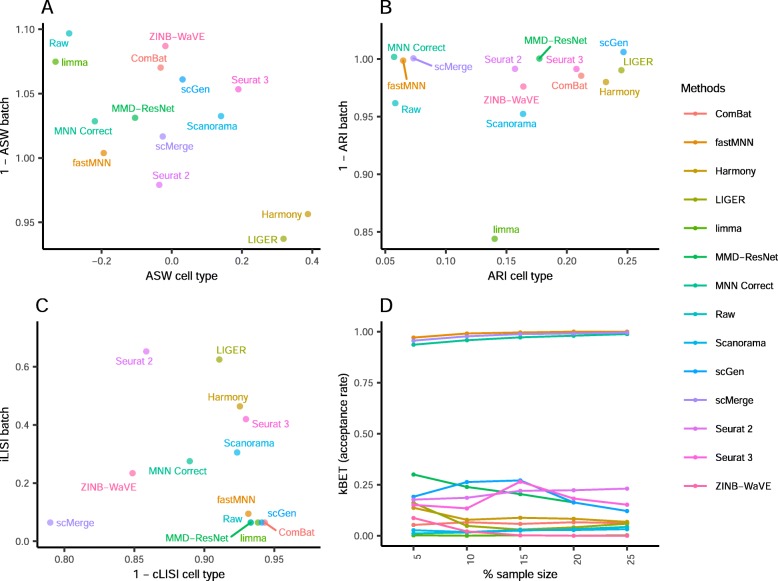
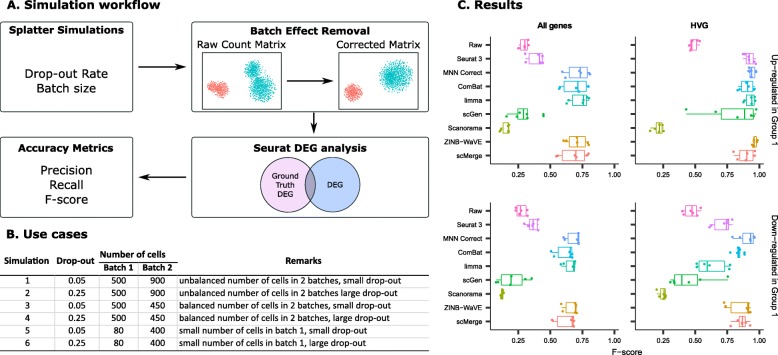
Results: We compare 14 methods in terms of computational runtime, the ability to handle large datasets, and batch-effect correction efficacy while preserving cell type purity. Five scenarios are designed for the study: identical cell types with different technologies, non-identical cell types, multiple batches, big data, and simulated data. Performance is evaluated using four benchmarking metrics including kBET, LISI, ASW, and ARI. We also investigate the use of batch-corrected data to study differential gene expression.
Conclusion: Based on our results, Harmony, LIGER, and Seurat 3 are the recommended methods for batch integration. Due to its significantly shorter runtime, Harmony is recommended as the first method to try, with the other methods as viable alternatives.
Keywords: Batch correction; Batch effect; Differential gene expression; Integration; Single-cell RNA-seq.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
















References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
