

Genomic programming of IRF4-expressing human Langerhans cells
- PMID: 31949143
- PMCID: PMC6965086
- DOI: 10.1038/s41467-019-14125-x
Genomic programming of IRF4-expressing human Langerhans cells
Abstract
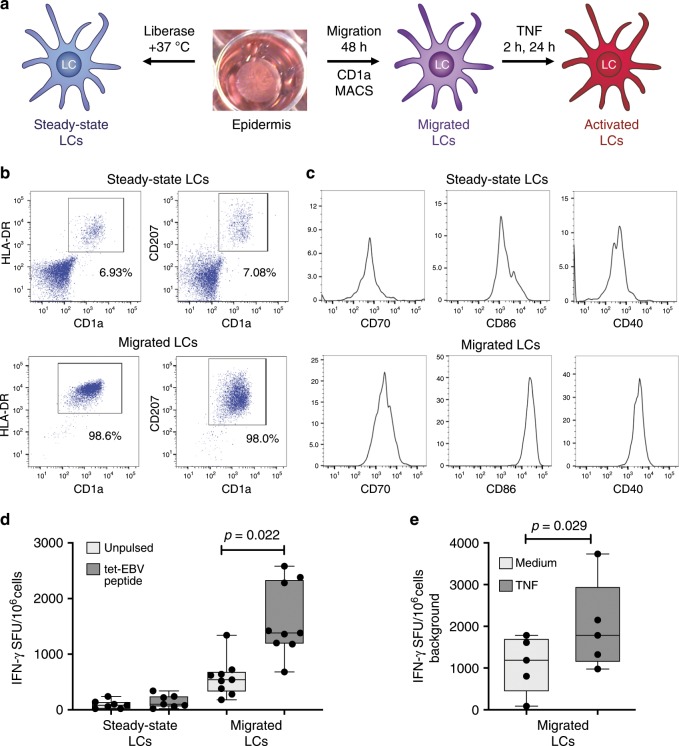
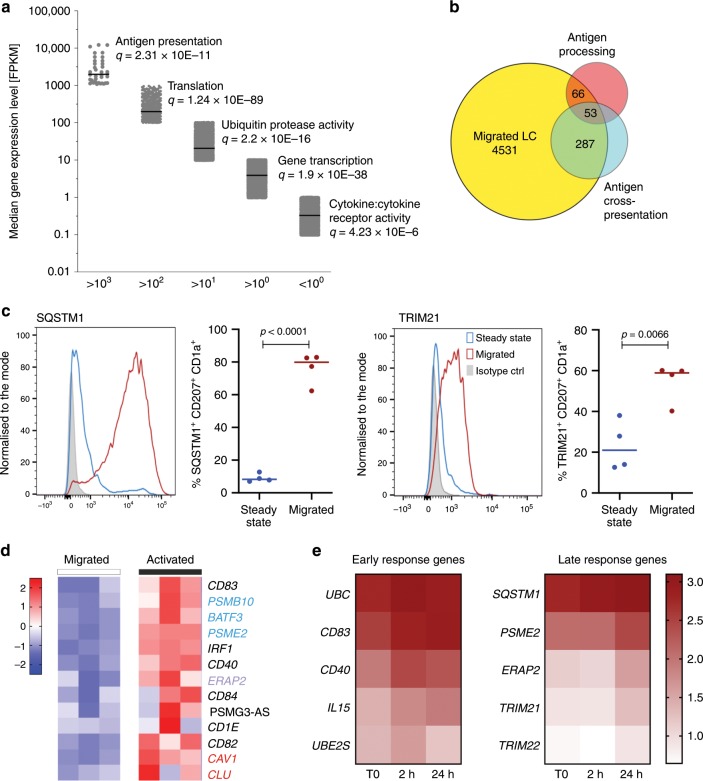
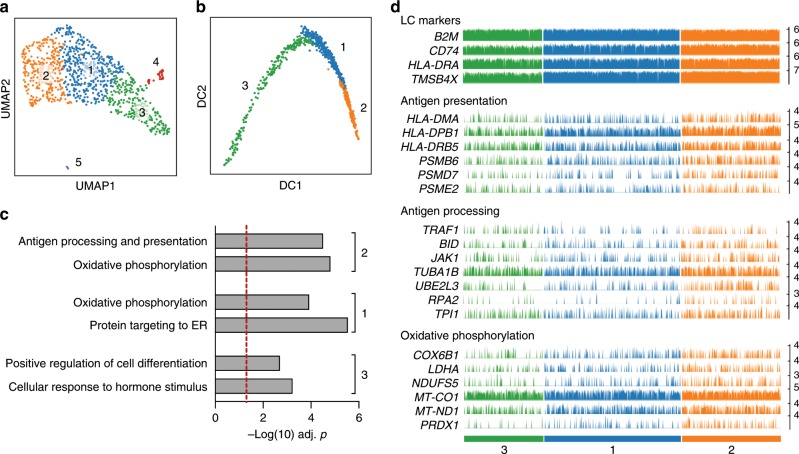
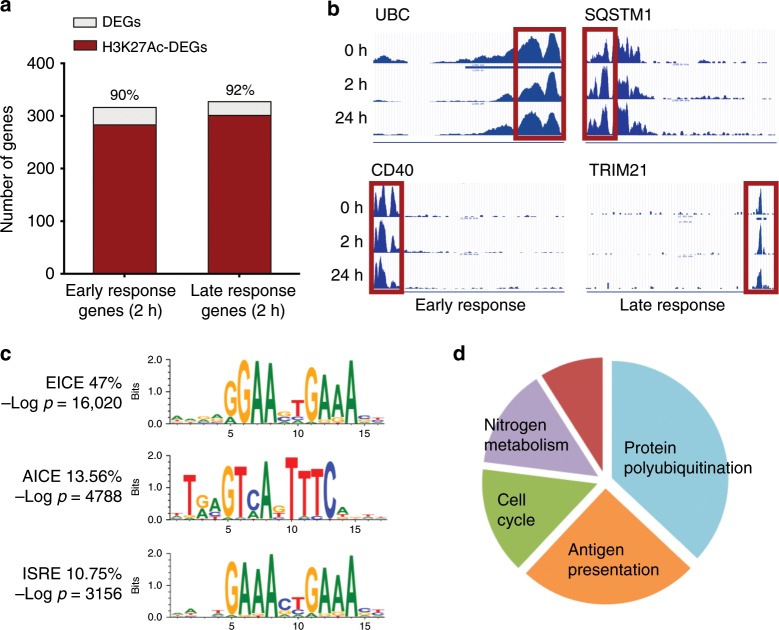
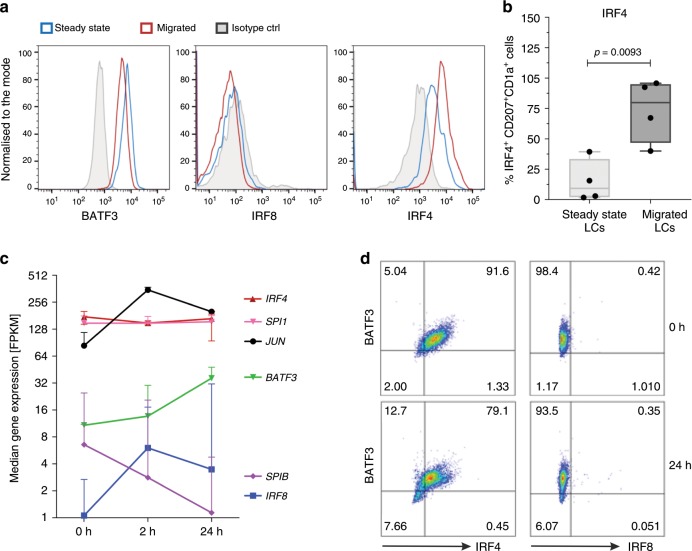
Langerhans cells (LC) can prime tolerogenic as well as immunogenic responses in skin, but the genomic states and transcription factors (TF) regulating these context-specific responses are unclear. Bulk and single-cell transcriptional profiling demonstrates that human migratory LCs are robustly programmed for MHC-I and MHC-II antigen presentation. Chromatin analysis reveals enrichment of ETS-IRF and AP1-IRF composite regulatory elements in antigen-presentation genes, coinciding with expression of the TFs, PU.1, IRF4 and BATF3 but not IRF8. Migration of LCs from the epidermis is accompanied by upregulation of IRF4, antigen processing components and co-stimulatory molecules. TNF stimulation augments LC cross-presentation while attenuating IRF4 expression. CRISPR-mediated editing reveals IRF4 to positively regulate the LC activation programme, but repress NF2EL2 and NF-kB pathway genes that promote responsiveness to oxidative stress and inflammatory cytokines. Thus, IRF4-dependent genomic programming of human migratory LCs appears to enable LC maturation while attenuating excessive inflammatory and immunogenic responses in the epidermis.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
