

Extreme intratumour heterogeneity and driver evolution in mismatch repair deficient gastro-oesophageal cancer
- PMID: 31949146
- PMCID: PMC6965135
- DOI: 10.1038/s41467-019-13915-7
Extreme intratumour heterogeneity and driver evolution in mismatch repair deficient gastro-oesophageal cancer
Erratum in
-
Author Correction: Extreme intratumour heterogeneity and driver evolution in mismatch repair deficient gastro-oesophageal cancer.Nat Commun. 2020 Jan 29;11(1):675. doi: 10.1038/s41467-020-14602-8. Nat Commun. 2020. PMID: 31996672 Free PMC article.
Abstract
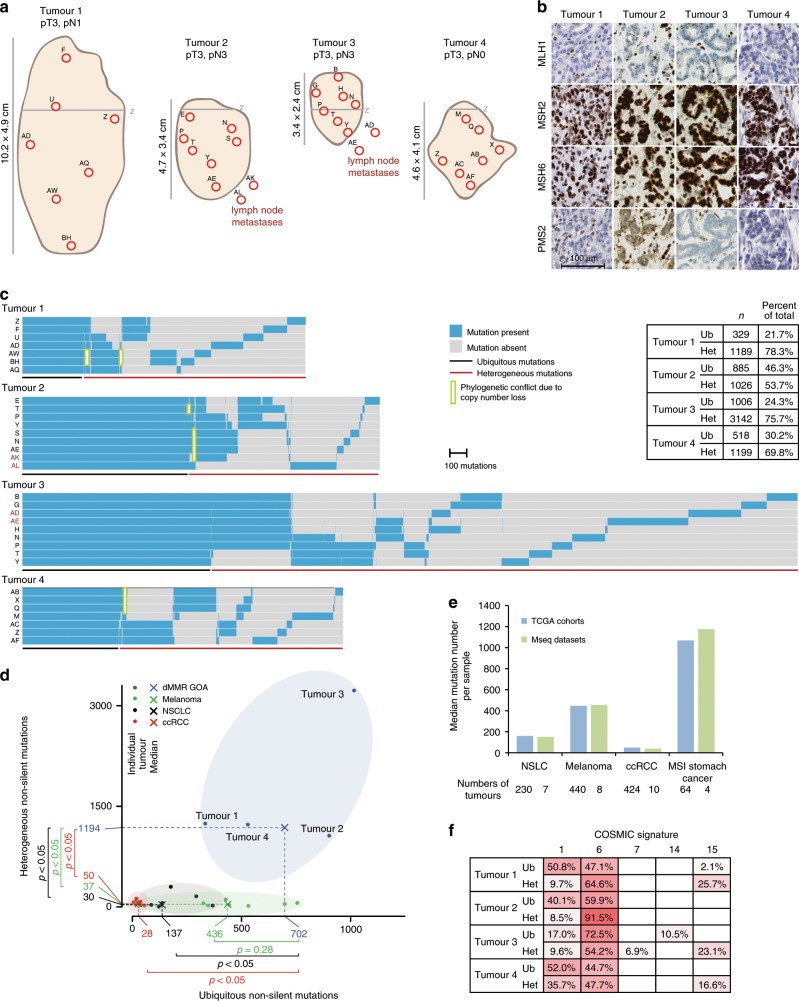
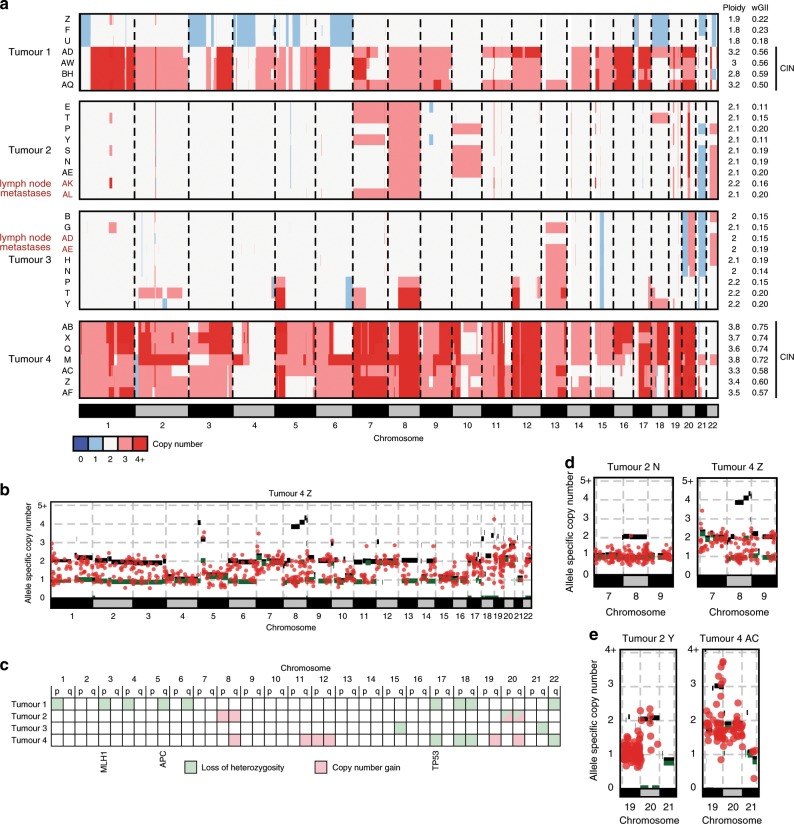
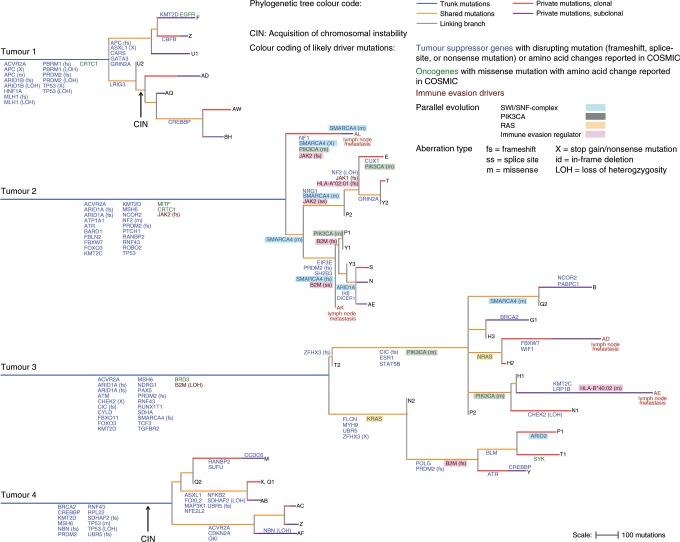
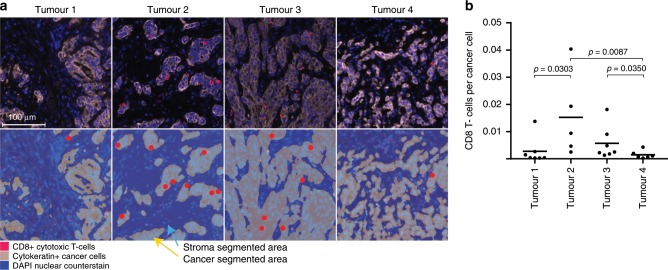
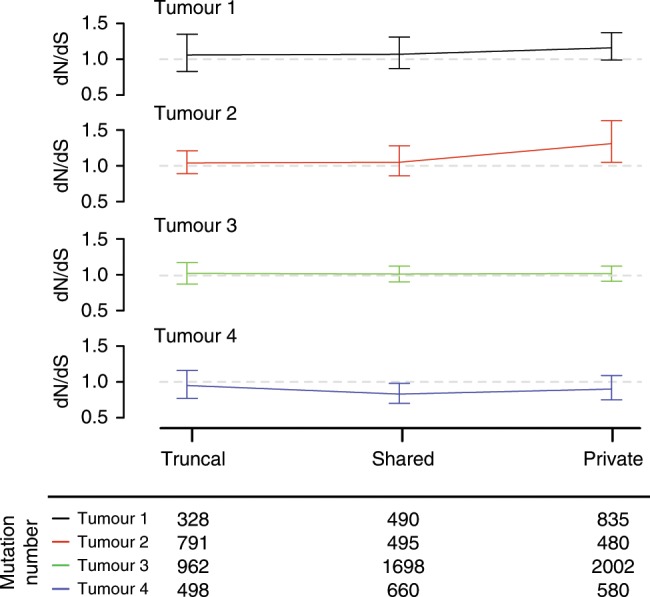
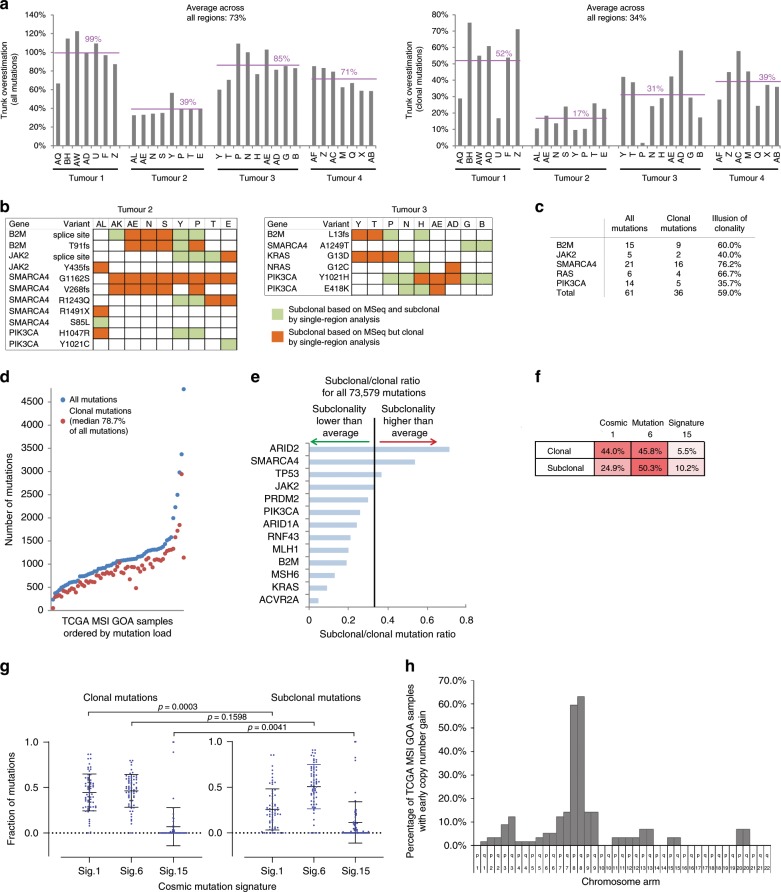
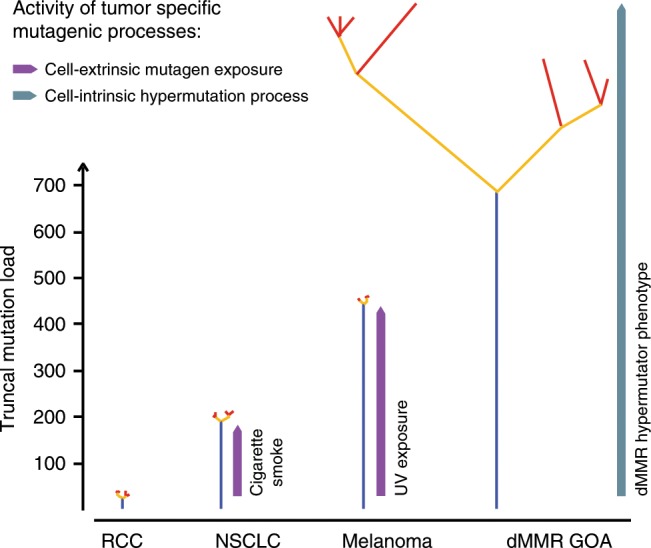
Mismatch repair deficient (dMMR) gastro-oesophageal adenocarcinomas (GOAs) show better outcomes than their MMR-proficient counterparts and high immunotherapy sensitivity. The hypermutator-phenotype of dMMR tumours theoretically enables high evolvability but their evolution has not been investigated. Here we apply multi-region exome sequencing (MSeq) to four treatment-naive dMMR GOAs. This reveals extreme intratumour heterogeneity (ITH), exceeding ITH in other cancer types >20-fold, but also long phylogenetic trunks which may explain the exquisite immunotherapy sensitivity of dMMR tumours. Subclonal driver mutations are common and parallel evolution occurs in RAS, PIK3CA, SWI/SNF-complex genes and in immune evasion regulators. MSeq data and evolution analysis of single region-data from 64 MSI GOAs show that chromosome 8 gains are early genetic events and that the hypermutator-phenotype remains active during progression. MSeq may be necessary for biomarker development in these heterogeneous cancers. Comparison with other MSeq-analysed tumour types reveals mutation rates and their timing to determine phylogenetic tree morphologies.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Ferlay J, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136:E359–E386. - PubMed
-
- Polom K, et al. Meta-analysis of microsatellite instability in relation to clinicopathological characteristics and overall survival in gastric cancer. Br. J. Surg. 2018;105:159–167. - PubMed
-
- Smyth Elizabeth Catherine, Hulkki Wilson Sanna, Nankivell Matthew Guy, Gonzalez de Castro David, Wotherspoon Andrew, Okines Alicia Frances Clare, Langley Ruth E, Stenning Sally Patricia, Cunningham David. Prognostic and predictive effect of microsatellite instability (MSI) in MAGIC. Journal of Clinical Oncology. 2015;33(3_suppl):62–62.
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
