

A restricted spectrum of missense KMT2D variants cause a multiple malformations disorder distinct from Kabuki syndrome
- PMID: 31949313
- PMCID: PMC7200597
- DOI: 10.1038/s41436-019-0743-3
A restricted spectrum of missense KMT2D variants cause a multiple malformations disorder distinct from Kabuki syndrome
Erratum in
-
Correction: A restricted spectrum of missense KMT2D variants cause a multiple malformations disorder distinct from Kabuki syndrome.Genet Med. 2020 May;22(5):980. doi: 10.1038/s41436-020-0784-7. Genet Med. 2020. PMID: 32203228 Free PMC article.
Abstract
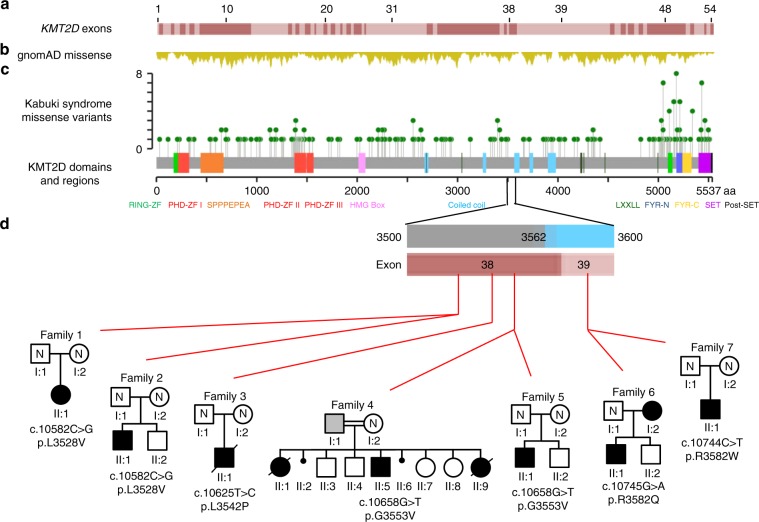
Purpose: To investigate if specific exon 38 or 39 KMT2D missense variants (MVs) cause a condition distinct from Kabuki syndrome type 1 (KS1).
Methods: Multiple individuals, with MVs in exons 38 or 39 of KMT2D that encode a highly conserved region of 54 amino acids flanked by Val3527 and Lys3583, were identified and phenotyped. Functional tests were performed to study their pathogenicity and understand the disease mechanism.
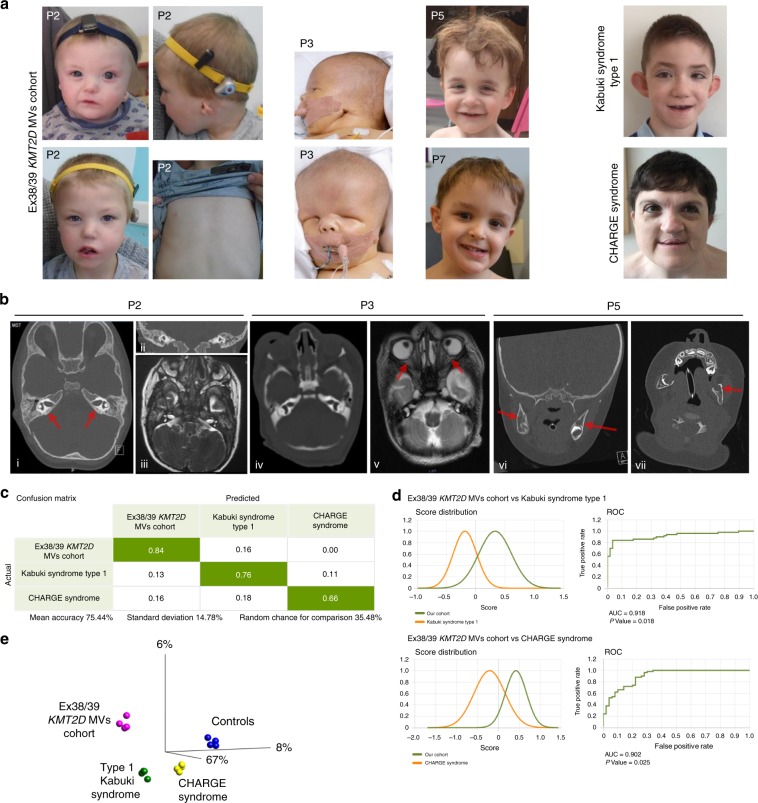
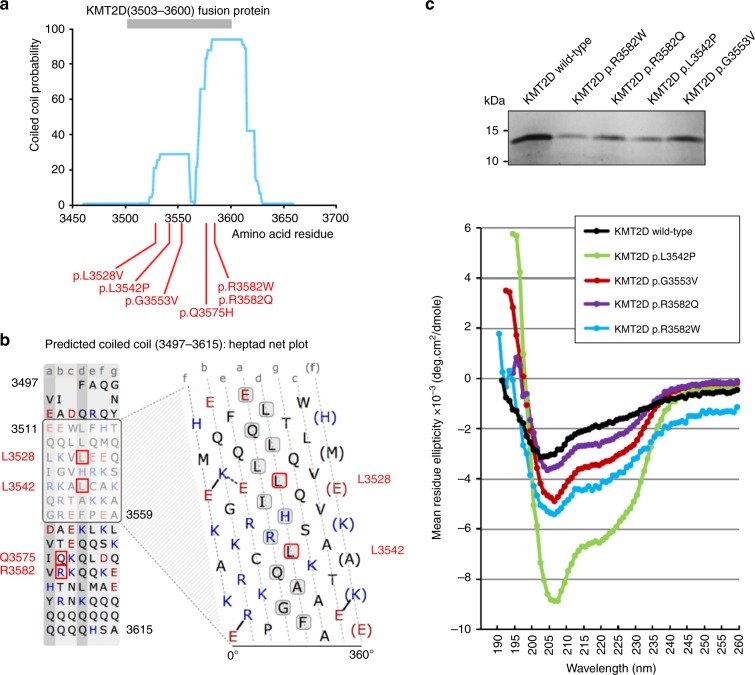
Results: The consistent clinical features of the affected individuals, from seven unrelated families, included choanal atresia, athelia or hypoplastic nipples, branchial sinus abnormalities, neck pits, lacrimal duct anomalies, hearing loss, external ear malformations, and thyroid abnormalities. None of the individuals had intellectual disability. The frequency of clinical features, objective software-based facial analysis metrics, and genome-wide peripheral blood DNA methylation patterns in these patients were significantly different from that of KS1. Circular dichroism spectroscopy indicated that these MVs perturb KMT2D secondary structure through an increased disordered to ɑ-helical transition.
Conclusion: KMT2D MVs located in a specific region spanning exons 38 and 39 and affecting highly conserved residues cause a novel multiple malformations syndrome distinct from KS1. Unlike KMT2D haploinsufficiency in KS1, these MVs likely result in disease through a dominant negative mechanism.
Keywords: KMT2D; Kabuki syndrome; histone 3 lysine 4 methyltransferase; intrinsically disordered region; multiple congenital anomaly.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
