GATAD2B-associated neurodevelopmental disorder (GAND): clinical and molecular insights into a NuRD-related disorder
- PMID: 31949314
- PMCID: PMC7920571
- DOI: 10.1038/s41436-019-0747-z
GATAD2B-associated neurodevelopmental disorder (GAND): clinical and molecular insights into a NuRD-related disorder
Erratum in
-
Correction: GATAD2B-associated neurodevelopmental disorder (GAND): clinical and molecular insights into a NuRD-related disorder.Genet Med. 2020 Apr;22(4):822. doi: 10.1038/s41436-020-0760-2. Genet Med. 2020. PMID: 32047287 Free PMC article.
Abstract
Purpose: Determination of genotypic/phenotypic features of GATAD2B-associated neurodevelopmental disorder (GAND).
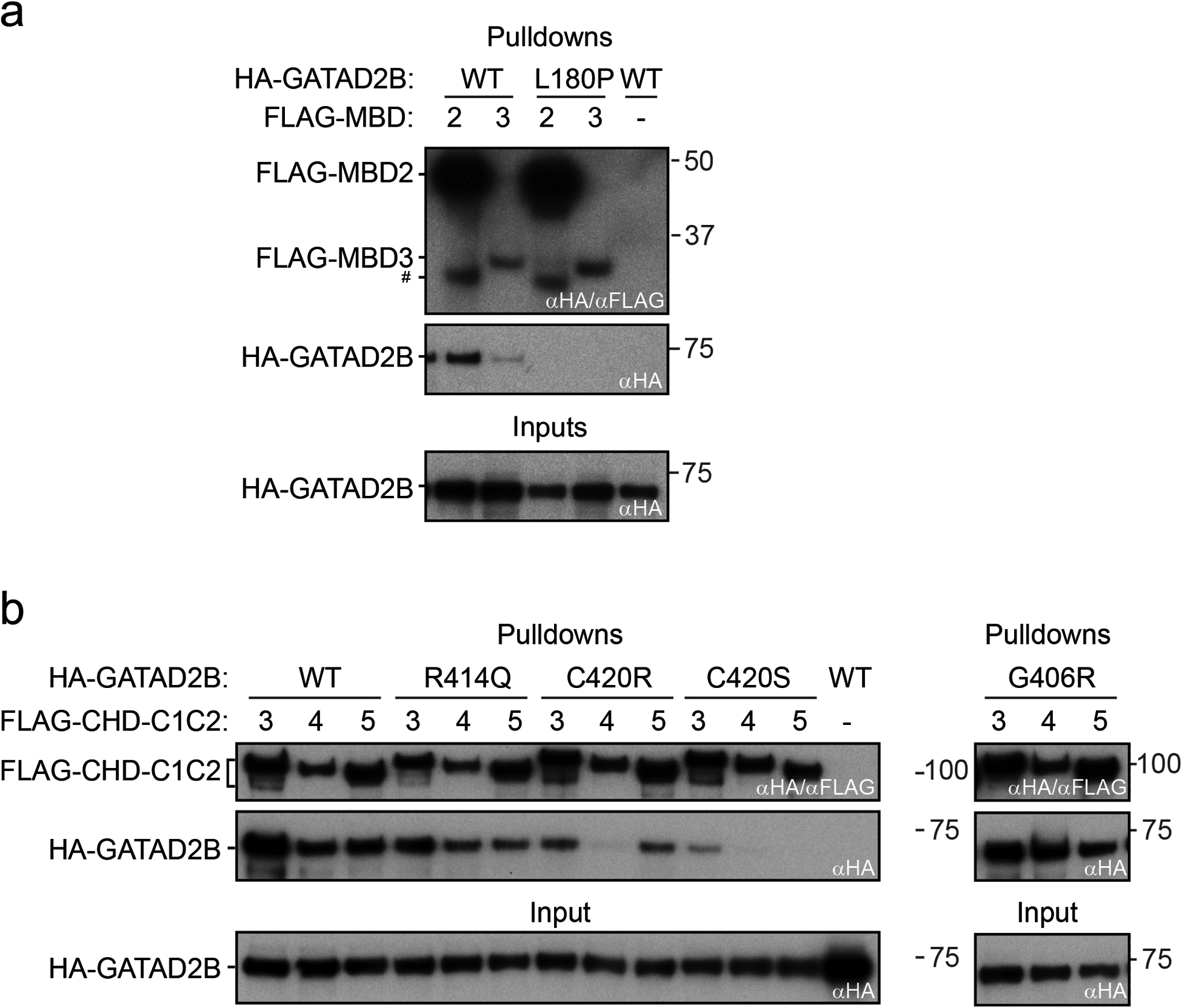
Methods: Fifty GAND subjects were evaluated to determine consistent genotypic/phenotypic features. Immunoprecipitation assays utilizing in vitro transcription-translation products were used to evaluate GATAD2B missense variants' ability to interact with binding partners within the nucleosome remodeling and deacetylase (NuRD) complex.
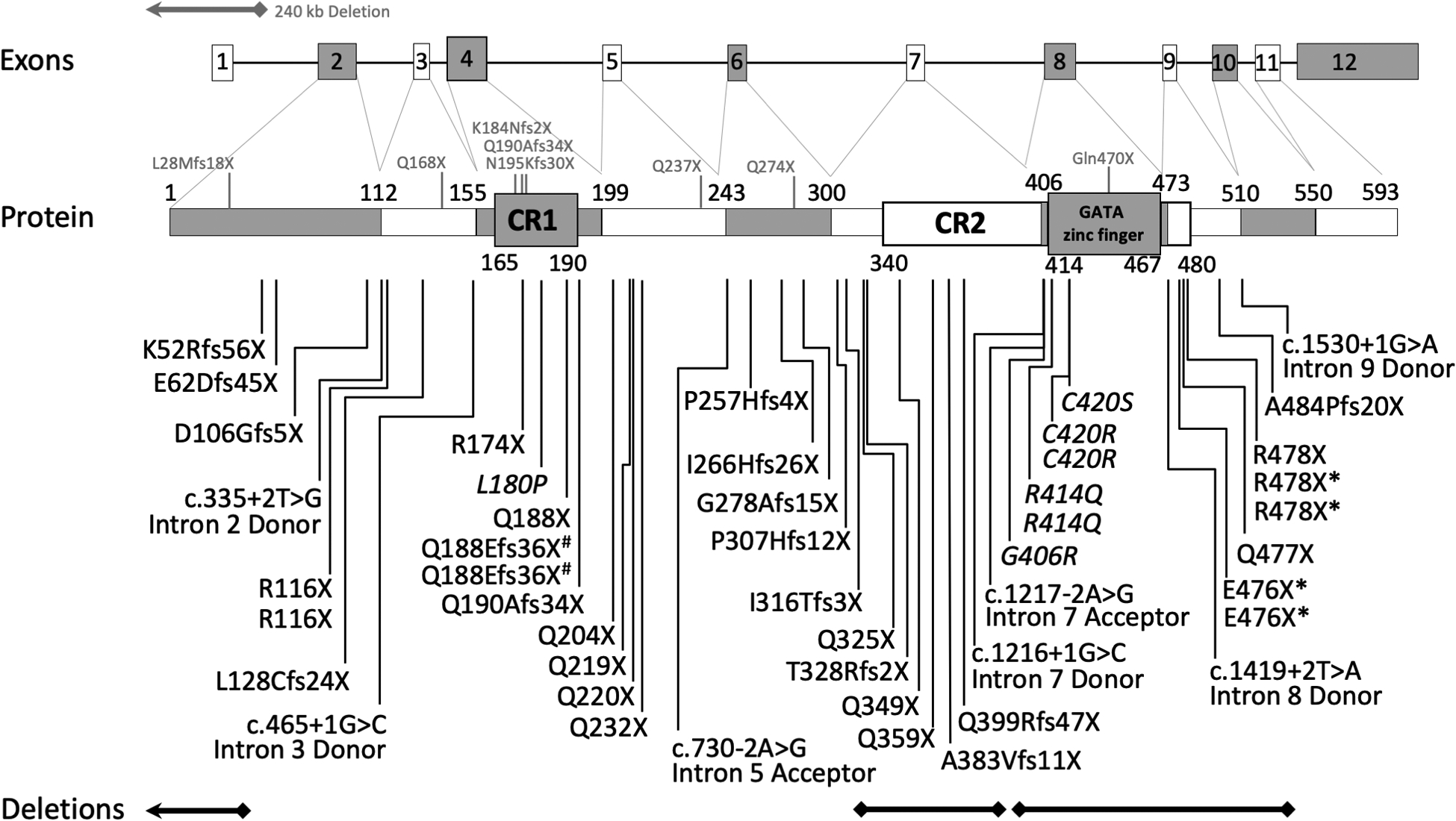
Results: Subjects had clinical findings that included macrocephaly, hypotonia, intellectual disability, neonatal feeding issues, polyhydramnios, apraxia of speech, epilepsy, and bicuspid aortic valves. Forty-one novelGATAD2B variants were identified with multiple variant types (nonsense, truncating frameshift, splice-site variants, deletions, and missense). Seven subjects were identified with missense variants that localized within two conserved region domains (CR1 or CR2) of the GATAD2B protein. Immunoprecipitation assays revealed several of these missense variants disrupted GATAD2B interactions with its NuRD complex binding partners.
Conclusions: A consistent GAND phenotype was caused by a range of genetic variants in GATAD2B that include loss-of-function and missense subtypes. Missense variants were present in conserved region domains that disrupted assembly of NuRD complex proteins. GAND's clinical phenotype had substantial clinical overlap with other disorders associated with the NuRD complex that involve CHD3 and CHD4, with clinical features of hypotonia, intellectual disability, cardiac defects, childhood apraxia of speech, and macrocephaly.
Keywords: GATAD2B; NuRD complex; apraxia of speech; chromatin remodeling; macrocephaly.
Conflict of interest statement
Figures