

Dating genomic variants and shared ancestry in population-scale sequencing data
- PMID: 31951611
- PMCID: PMC6992231
- DOI: 10.1371/journal.pbio.3000586
Dating genomic variants and shared ancestry in population-scale sequencing data
Abstract
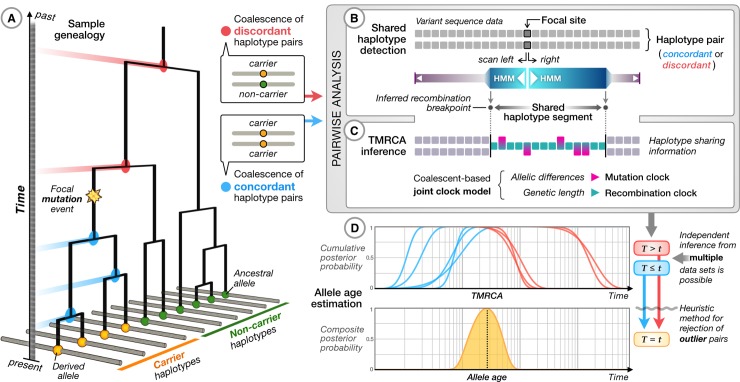
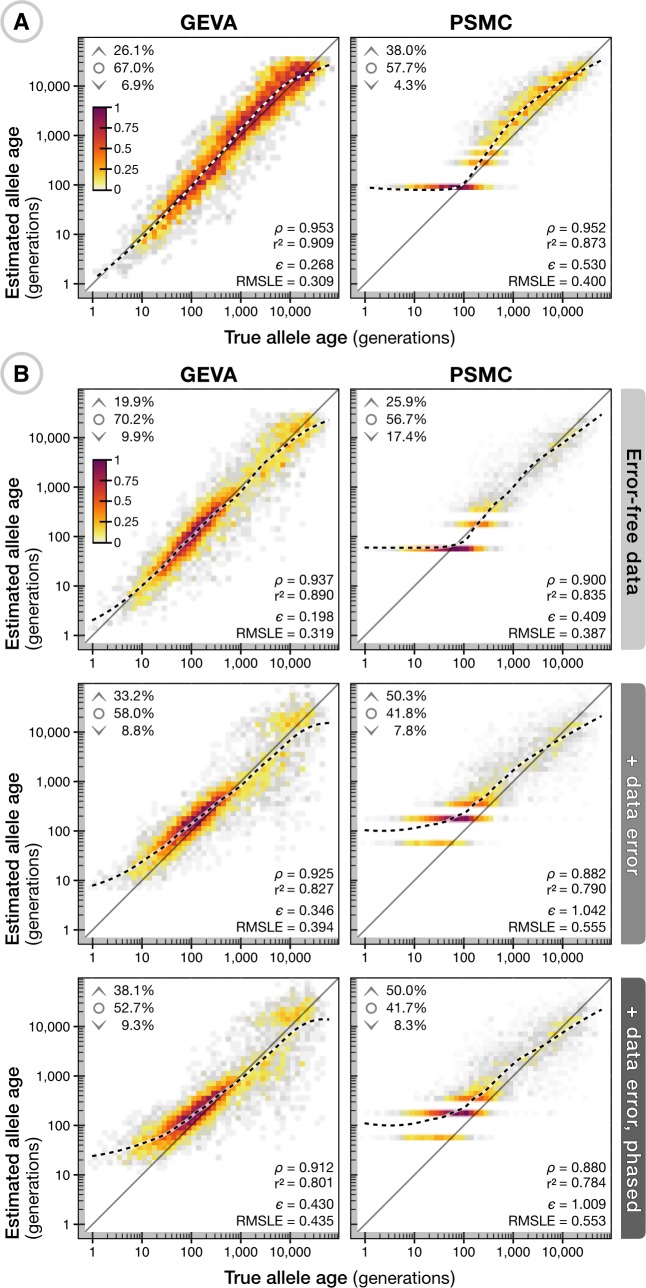
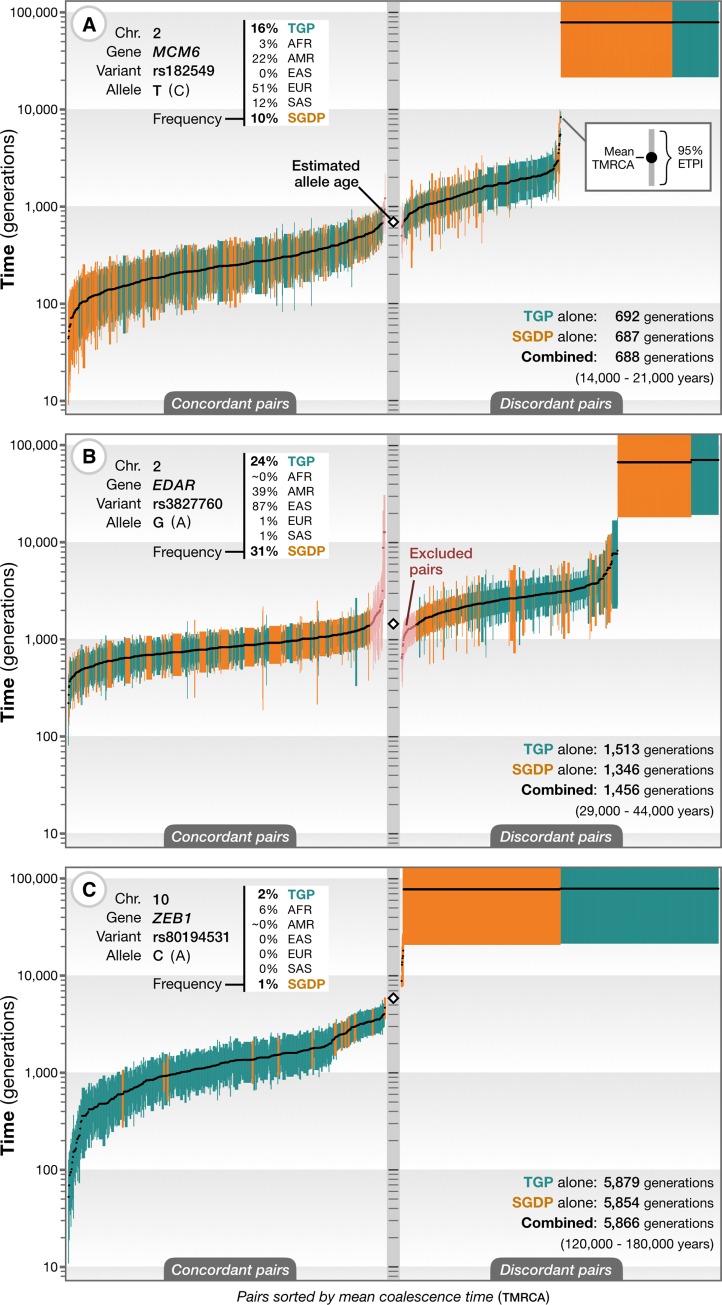
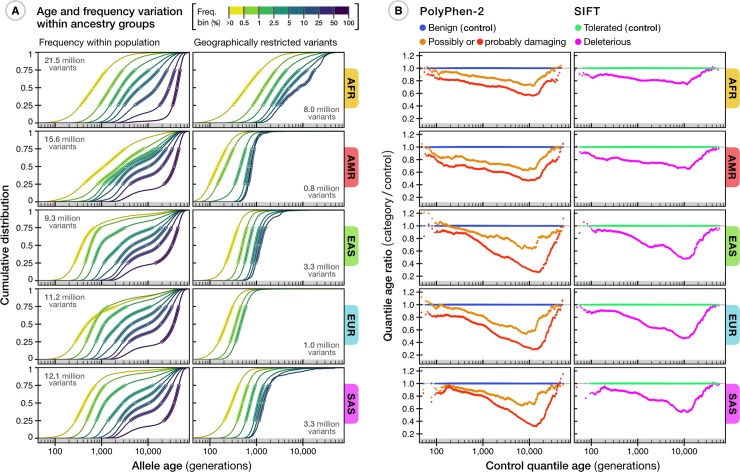
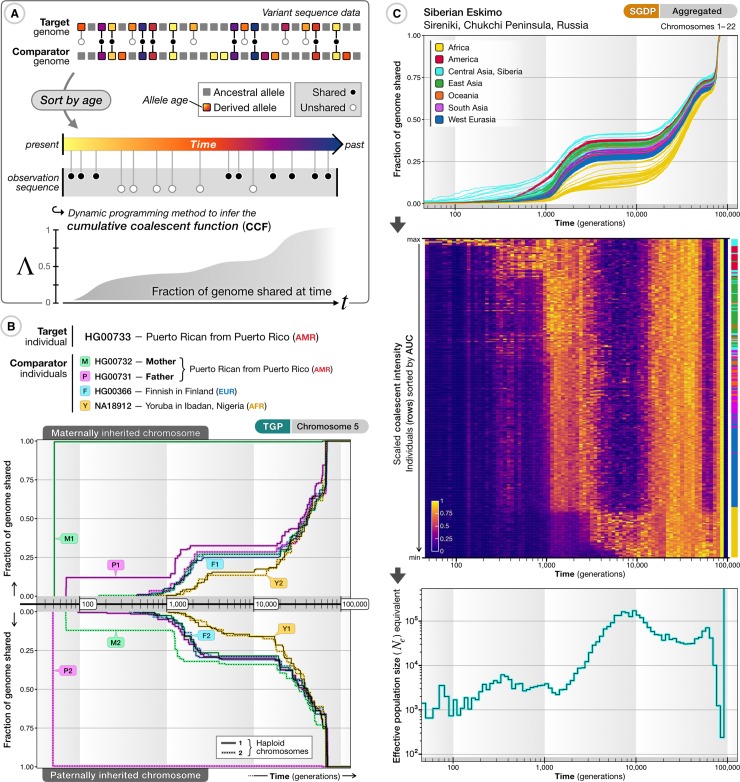
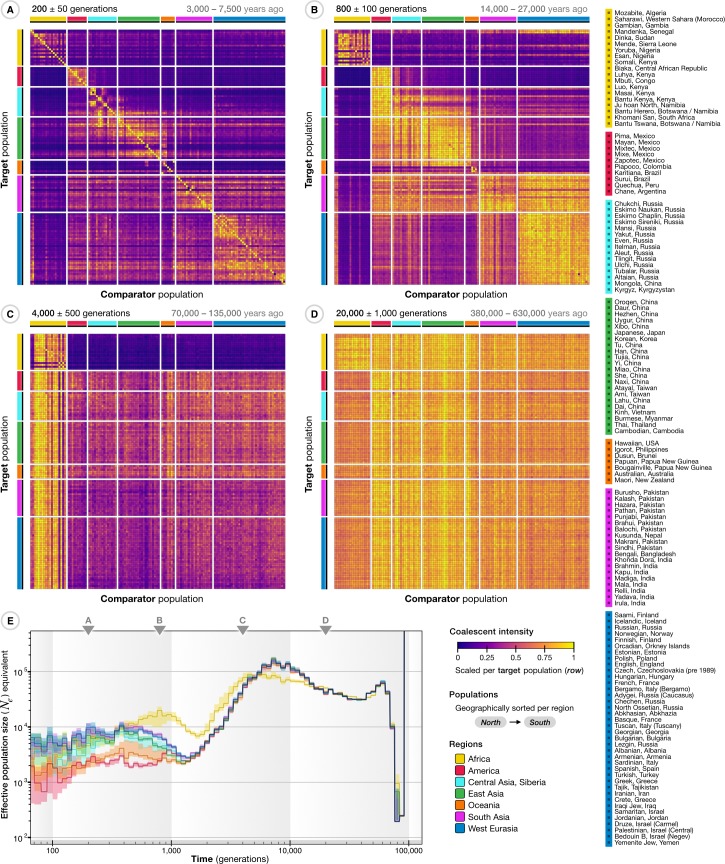
The origin and fate of new mutations within species is the fundamental process underlying evolution. However, while much attention has been focused on characterizing the presence, frequency, and phenotypic impact of genetic variation, the evolutionary histories of most variants are largely unexplored. We have developed a nonparametric approach for estimating the date of origin of genetic variants in large-scale sequencing data sets. The accuracy and robustness of the approach is demonstrated through simulation. Using data from two publicly available human genomic diversity resources, we estimated the age of more than 45 million single-nucleotide polymorphisms (SNPs) in the human genome and release the Atlas of Variant Age as a public online database. We characterize the relationship between variant age and frequency in different geographical regions and demonstrate the value of age information in interpreting variants of functional and selective importance. Finally, we use allele age estimates to power a rapid approach for inferring the ancestry shared between individual genomes and to quantify genealogical relationships at different points in the past, as well as to describe and explore the evolutionary history of modern human populations.
Conflict of interest statement
I have read the journal's policy and the authors of this manuscript have the following competing interests: GM is a shareholder in and non-executive director of Genomics PLC, and is a partner in Peptide Groove LLP. PKA is a shareholder in and a director of BioMe Oxford Ltd.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
