

Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia
- PMID: 31951650
- PMCID: PMC7059518
- DOI: 10.1182/blood.2019002779
Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia
Abstract
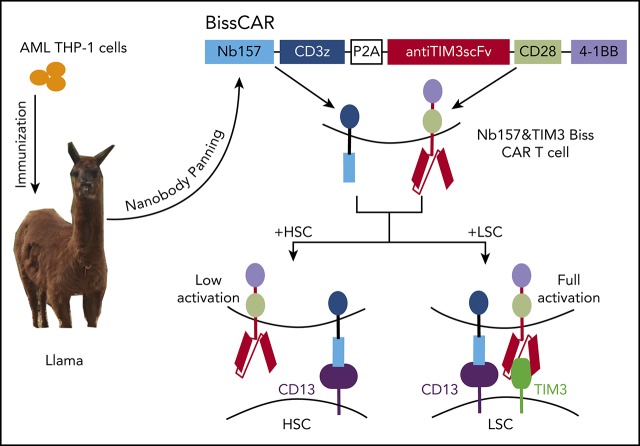
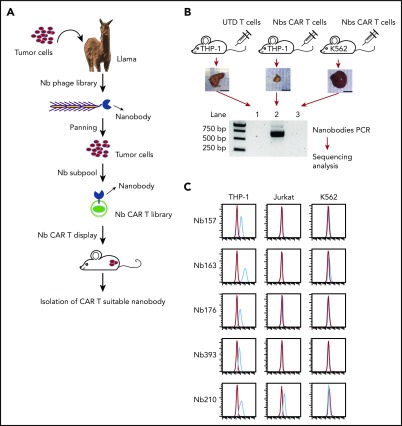
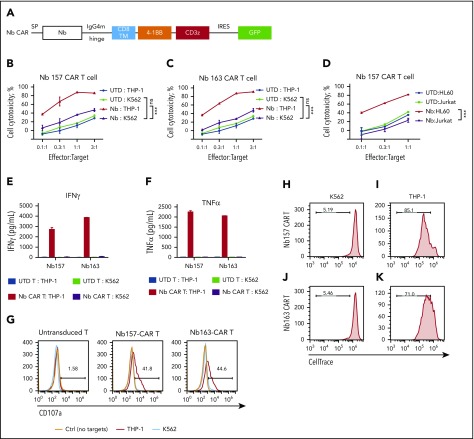
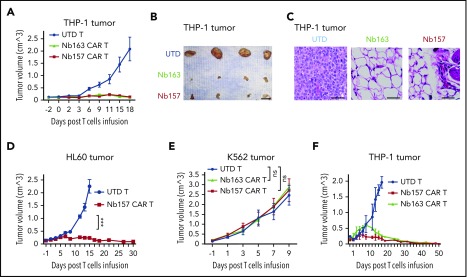
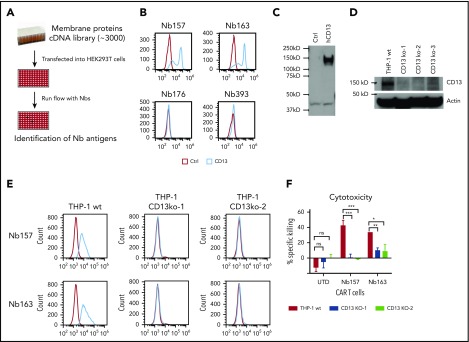
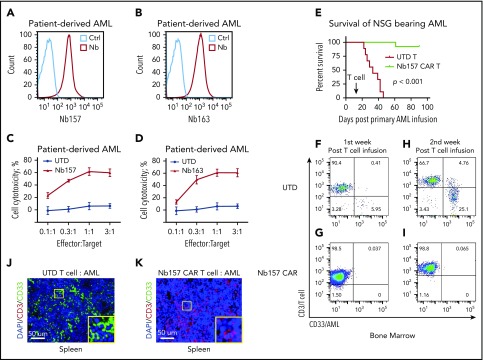
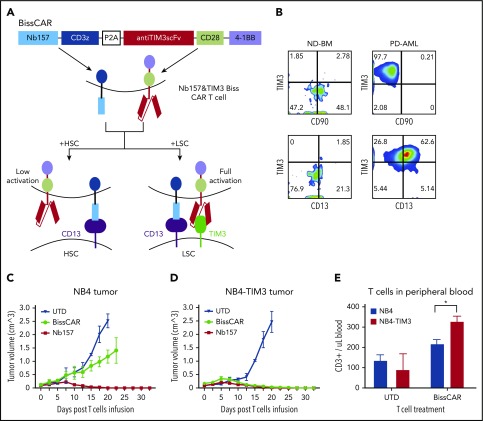
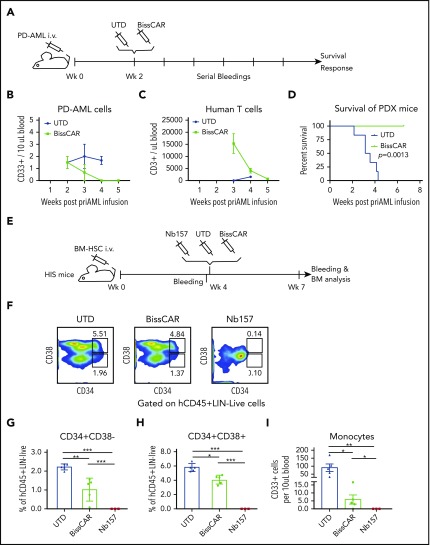
Chimeric antigen receptor (CAR) T cells have radically improved the treatment of B cell-derived malignancies by targeting CD19. The success has not yet expanded to treat acute myeloid leukemia (AML). We developed a Sequentially Tumor-Selected Antibody and Antigen Retrieval (STAR) system to rapidly isolate multiple nanobodies (Nbs) that preferentially bind AML cells and empower CAR T cells with anti-AML efficacy. STAR-isolated Nb157 specifically bound CD13, which is highly expressed in AML cells, and CD13 CAR T cells potently eliminated AML in vitro and in vivo. CAR T cells bispecific for CD13 and TIM3, which are upregulated in AML leukemia stem cells, eradicated patient-derived AML, with much reduced toxicity to human bone marrow stem cells and peripheral myeloid cells in mouse models, highlighting a promising approach for developing effective AML CAR T cell therapy.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
Comment in
-
A bispecific approach to improving CAR T cells in AML.Blood. 2020 Mar 5;135(10):703-704. doi: 10.1182/blood.2020004791. Blood. 2020. PMID: 32135017 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
