

Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy
- PMID: 31952359
- PMCID: PMC7017136
- DOI: 10.3390/genes11010099
Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy
Abstract
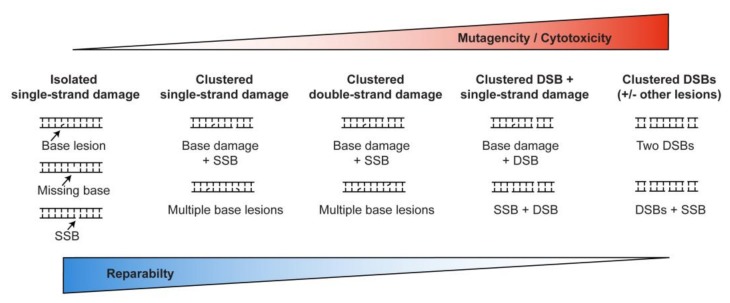
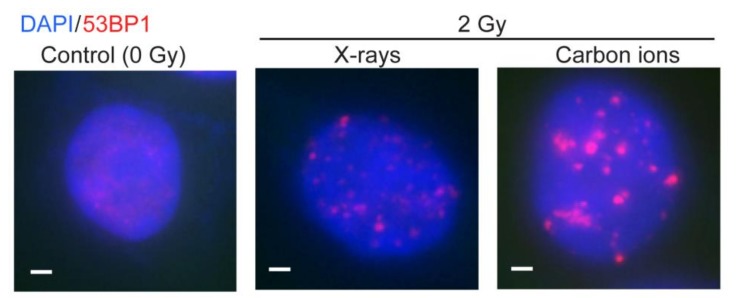
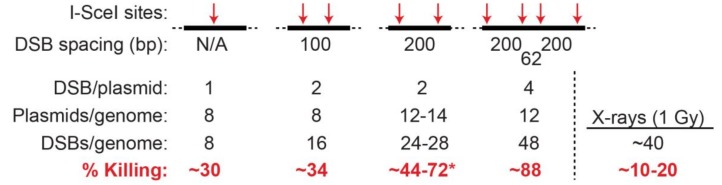
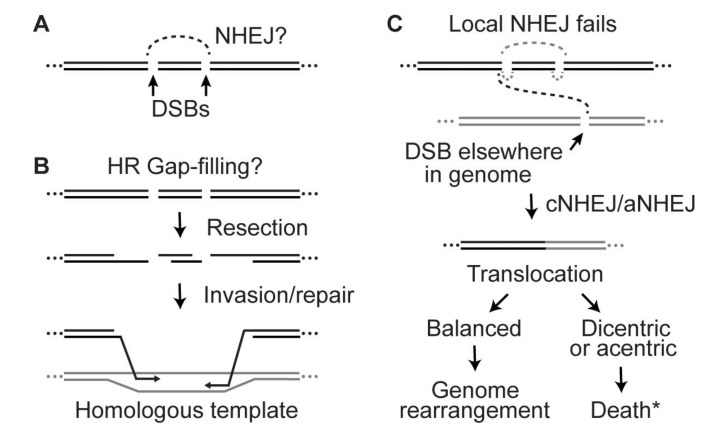
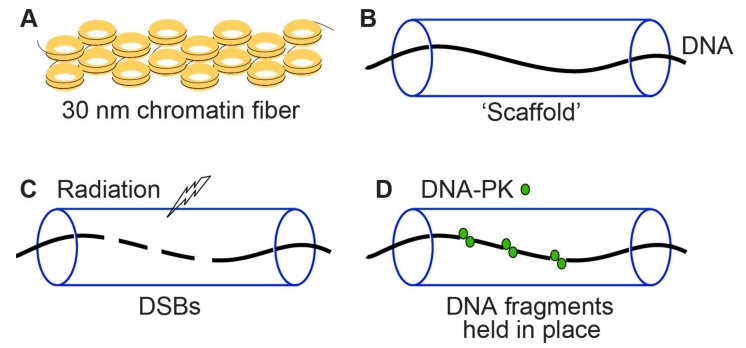
Cells manage to survive, thrive, and divide with high accuracy despite the constant threat of DNA damage. Cells have evolved with several systems that efficiently repair spontaneous, isolated DNA lesions with a high degree of accuracy. Ionizing radiation and a few radiomimetic chemicals can produce clustered DNA damage comprising complex arrangements of single-strand damage and DNA double-strand breaks (DSBs). There is substantial evidence that clustered DNA damage is more mutagenic and cytotoxic than isolated damage. Radiation-induced clustered DNA damage has proven difficult to study because the spectrum of induced lesions is very complex, and lesions are randomly distributed throughout the genome. Nonetheless, it is fairly well-established that radiation-induced clustered DNA damage, including non-DSB and DSB clustered lesions, are poorly repaired or fail to repair, accounting for the greater mutagenic and cytotoxic effects of clustered lesions compared to isolated lesions. High linear energy transfer (LET) charged particle radiation is more cytotoxic per unit dose than low LET radiation because high LET radiation produces more clustered DNA damage. Studies with I-SceI nuclease demonstrate that nuclease-induced DSB clusters are also cytotoxic, indicating that this cytotoxicity is independent of radiogenic lesions, including single-strand lesions and chemically "dirty" DSB ends. The poor repair of clustered DSBs at least in part reflects inhibition of canonical NHEJ by short DNA fragments. This shifts repair toward HR and perhaps alternative NHEJ, and can result in chromothripsis-mediated genome instability or cell death. These principals are important for cancer treatment by low and high LET radiation.
Keywords: DNA base damage; DNA double-strand breaks; chromatin; complex DNA lesions; cytotoxicity; genome instability; ionizing radiation; radiation oncology.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
