

Loss of centromere function drives karyotype evolution in closely related Malassezia species
- PMID: 31958060
- PMCID: PMC7025860
- DOI: 10.7554/eLife.53944
Loss of centromere function drives karyotype evolution in closely related Malassezia species
Abstract
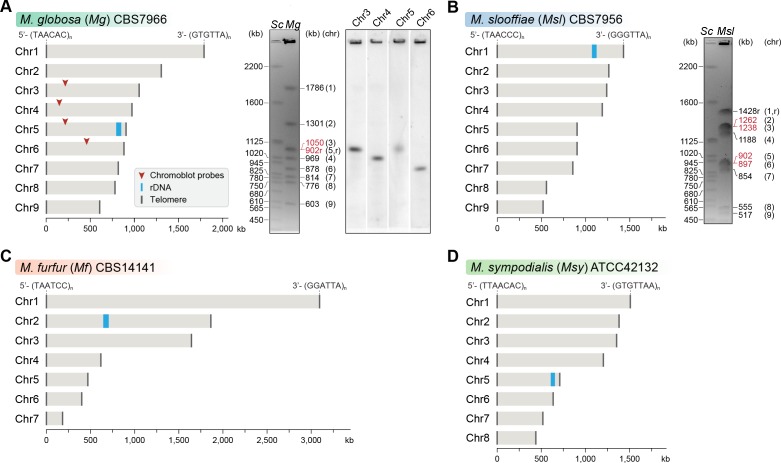
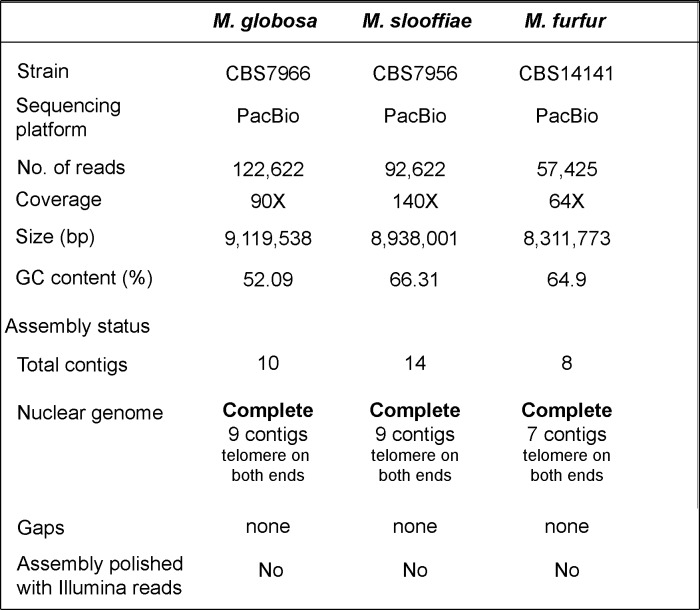
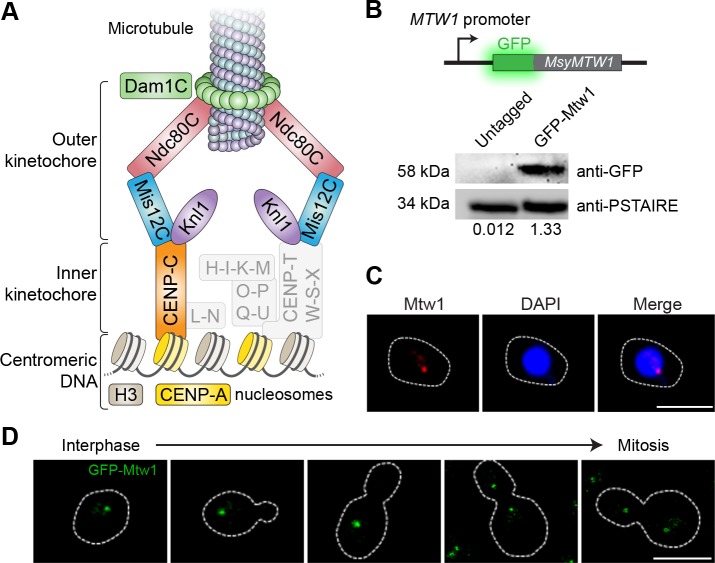
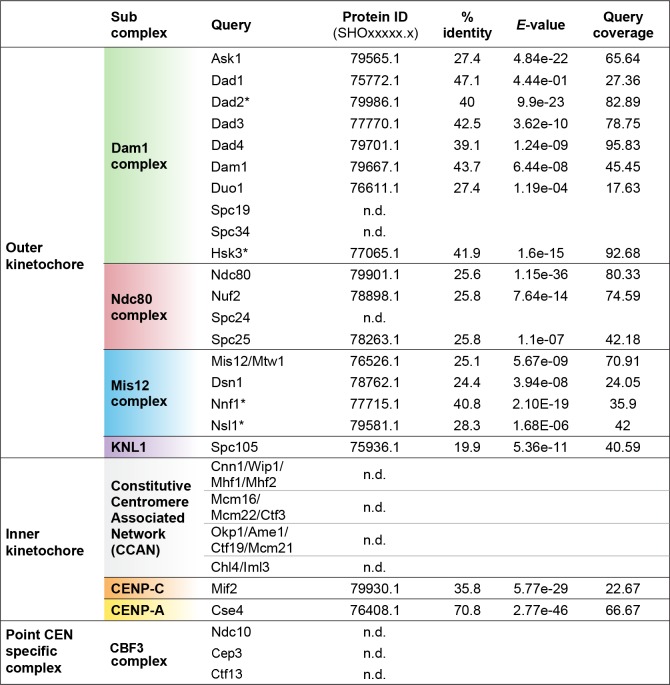
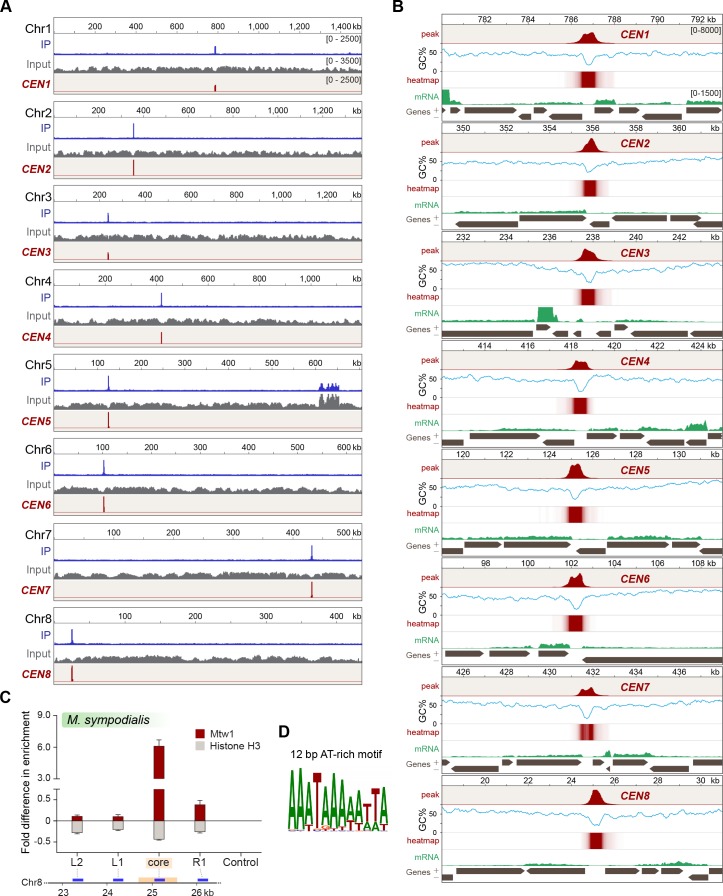
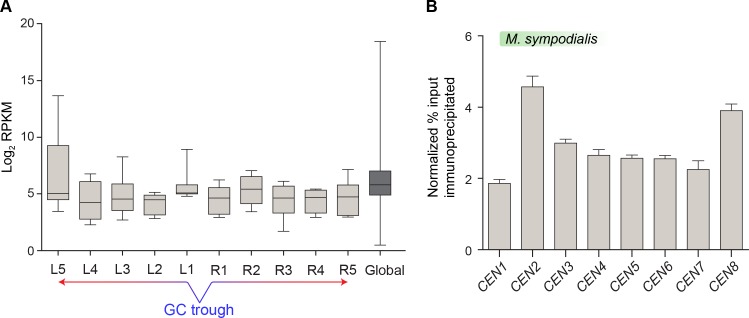
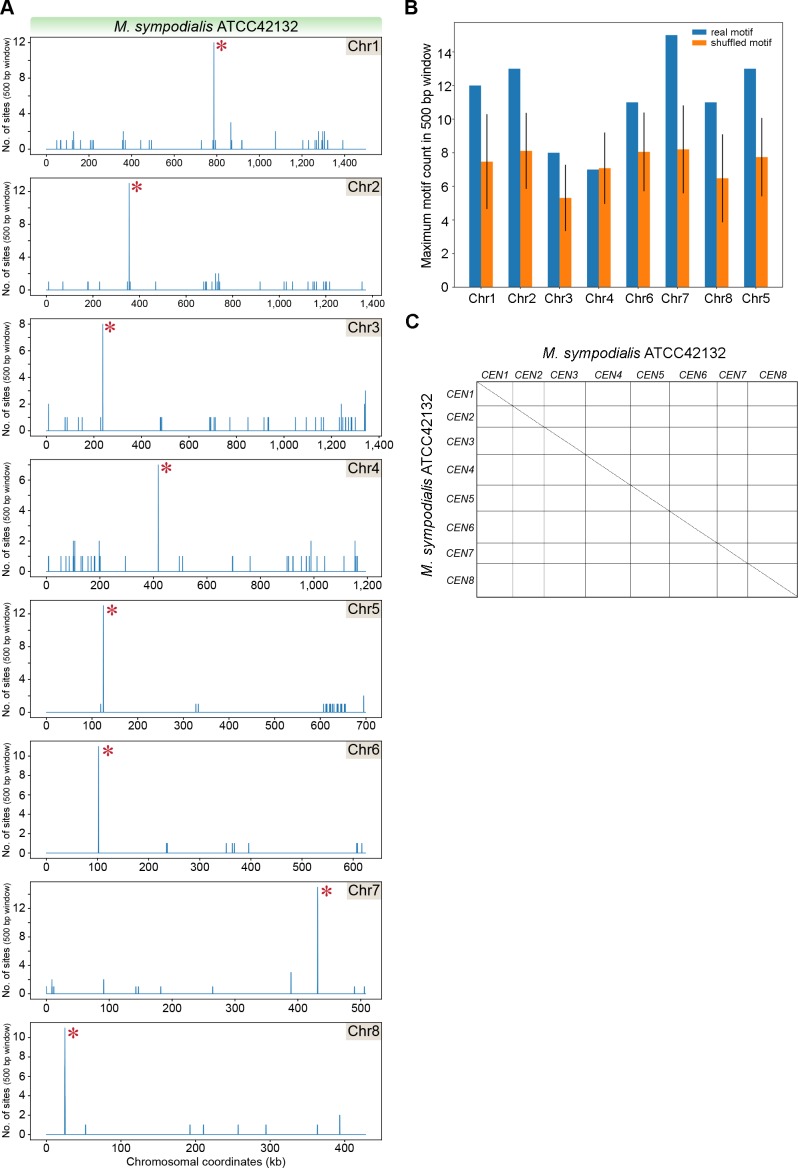
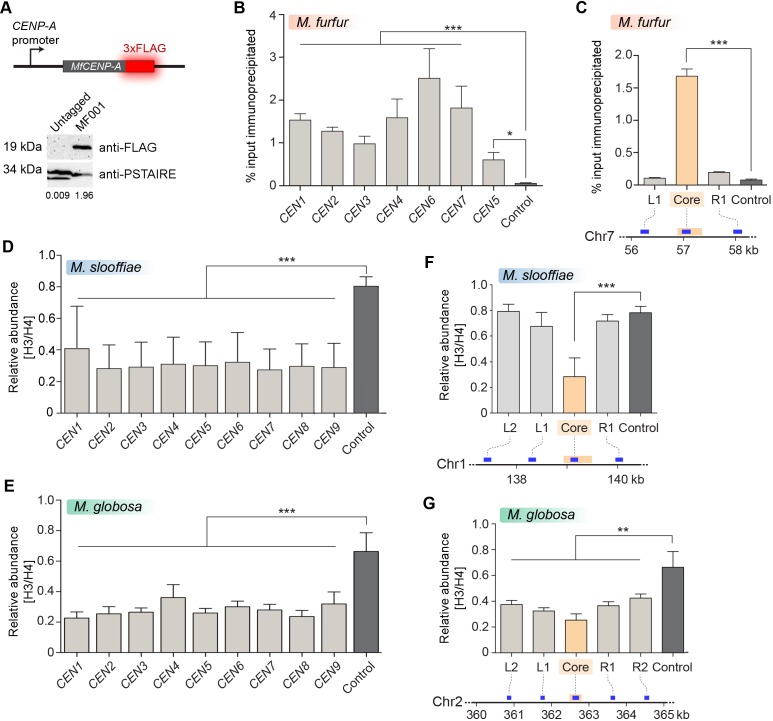
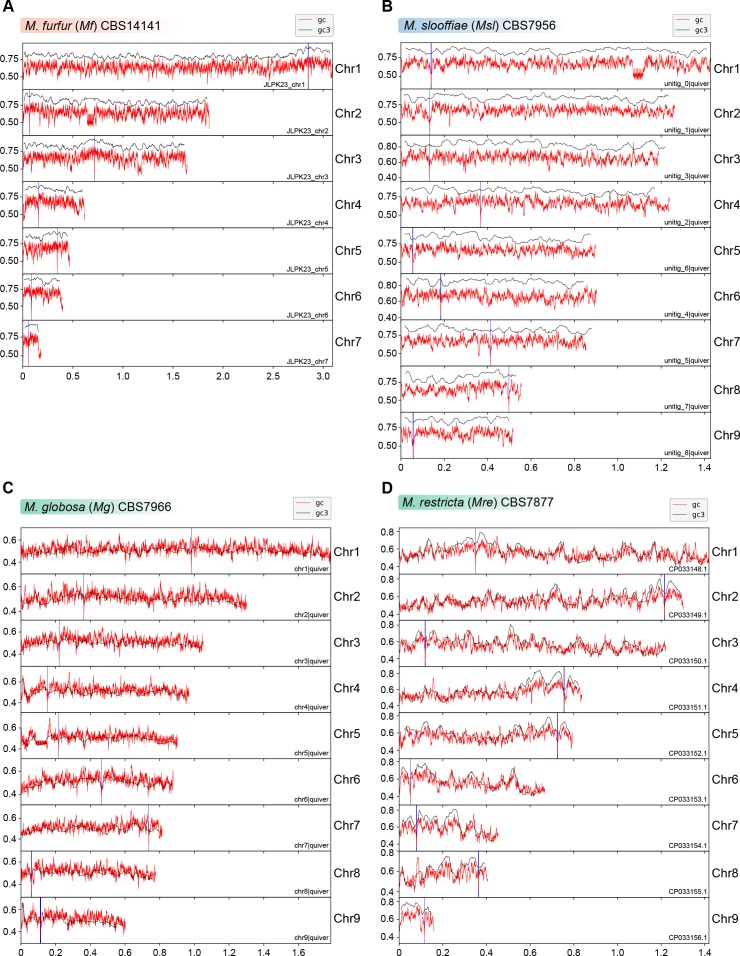
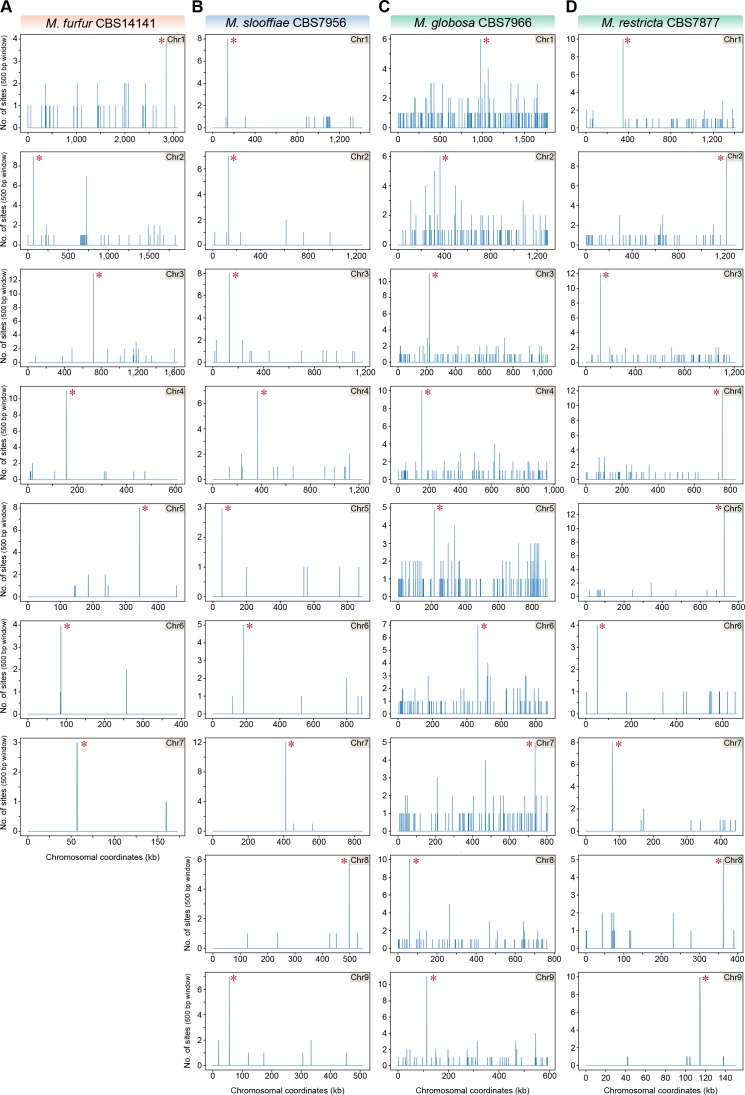
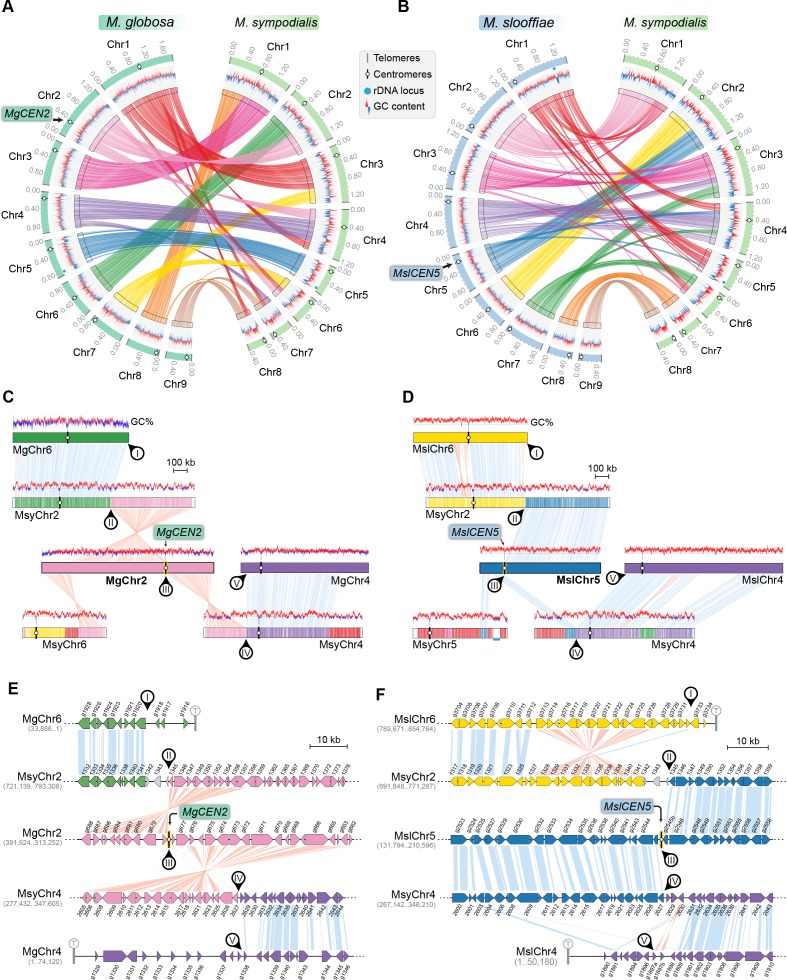
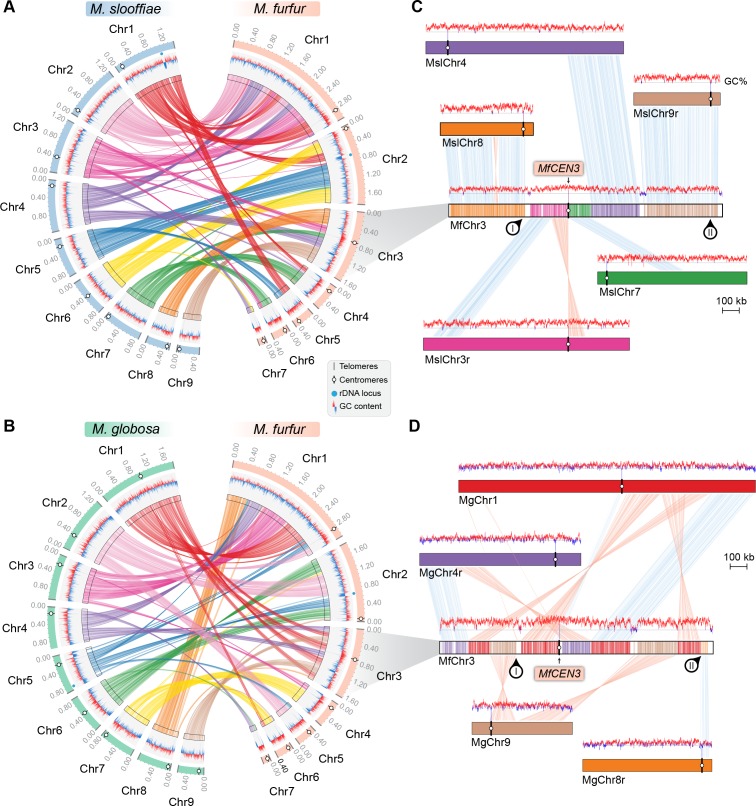
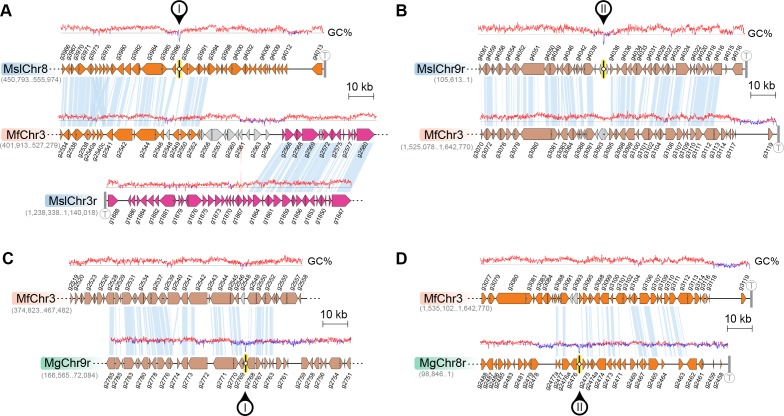
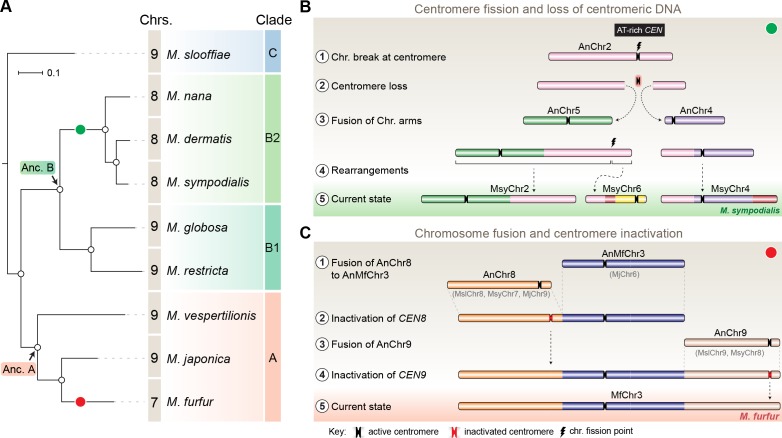
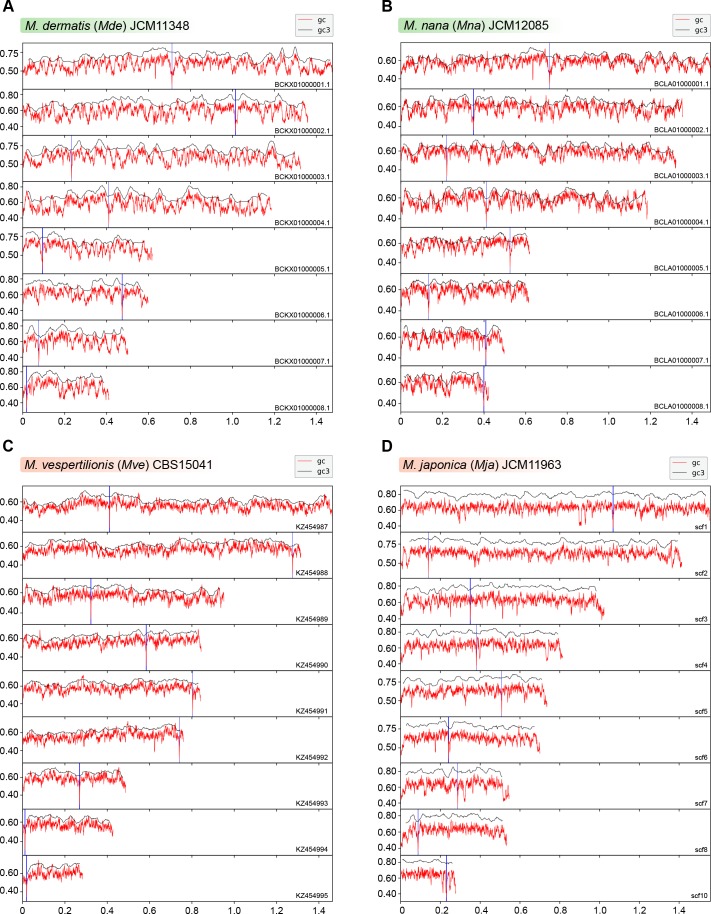
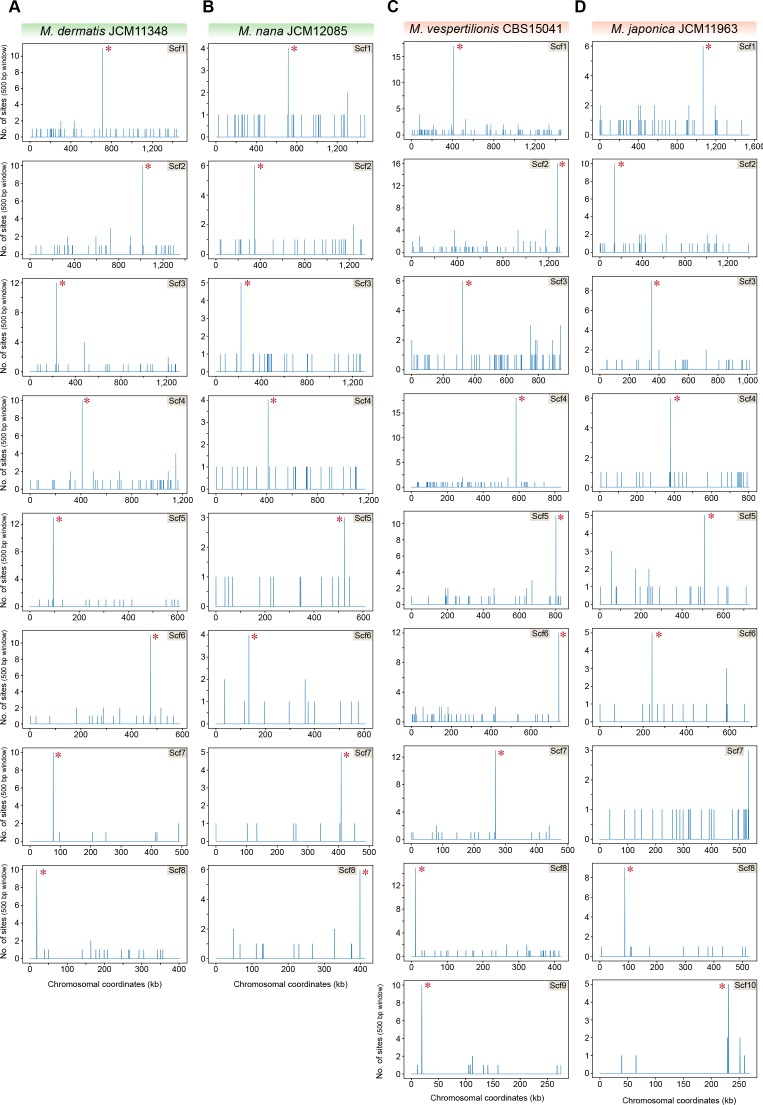
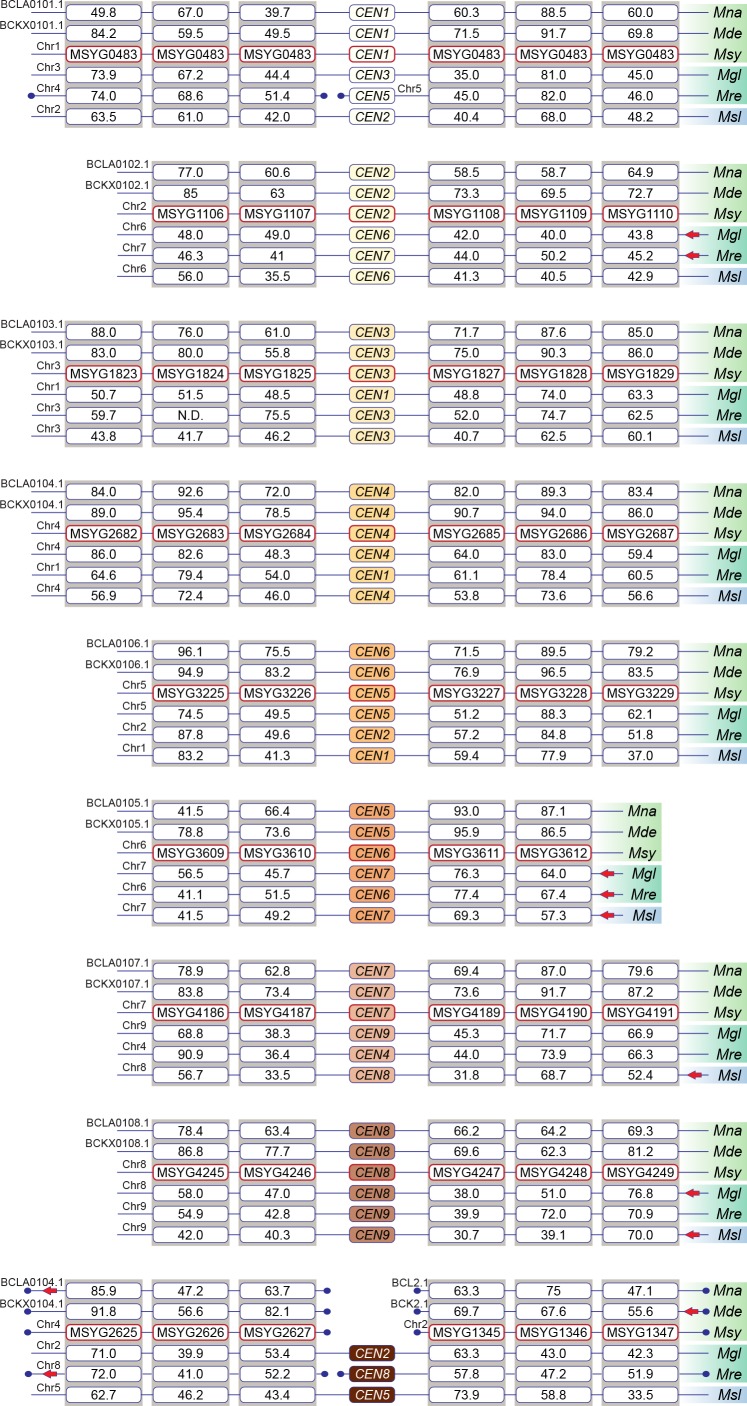
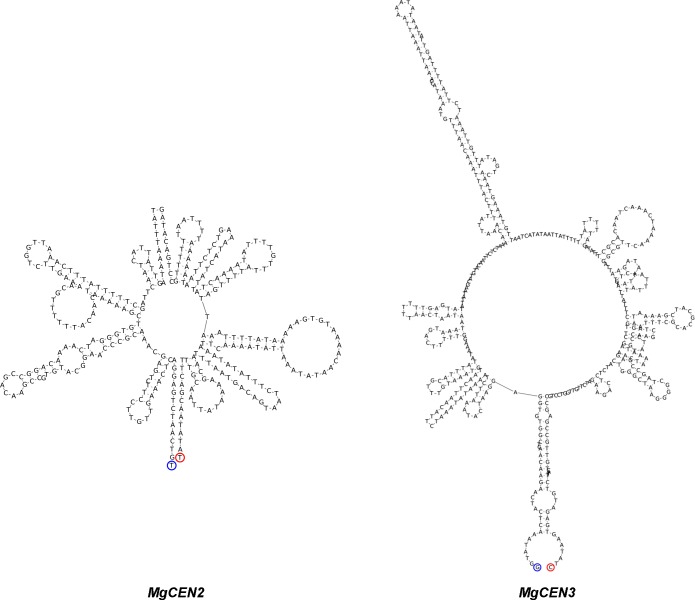
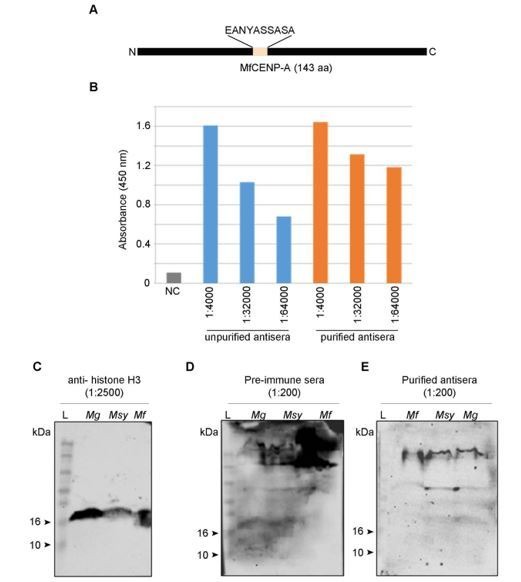
Genomic rearrangements associated with speciation often result in variation in chromosome number among closely related species. Malassezia species show variable karyotypes ranging between six and nine chromosomes. Here, we experimentally identified all eight centromeres in M. sympodialis as 3-5-kb long kinetochore-bound regions that span an AT-rich core and are depleted of the canonical histone H3. Centromeres of similar sequence features were identified as CENP-A-rich regions in Malassezia furfur, which has seven chromosomes, and histone H3 depleted regions in Malassezia slooffiae and Malassezia globosa with nine chromosomes each. Analysis of synteny conservation across centromeres with newly generated chromosome-level genome assemblies suggests two distinct mechanisms of chromosome number reduction from an inferred nine-chromosome ancestral state: (a) chromosome breakage followed by loss of centromere DNA and (b) centromere inactivation accompanied by changes in DNA sequence following chromosome-chromosome fusion. We propose that AT-rich centromeres drive karyotype diversity in the Malassezia species complex through breakage and inactivation.
Keywords: chromosome fusion; dicentric; double-strand breaks; genetics; genomics; kinetochore; skin microbe.
Plain language summary
Millions of yeast, bacteria and other microbes live in or on the human body. A type of yeast known as Malassezia is one of the most abundantmicrobes living on our skin. Generally, Malassezia do not cause symptoms in humans but are associated with dandruff, dermatitis and other skin conditions in susceptible individuals. They have also been found in the human gut, where they exacerbate Crohn’s disease and pancreatic cancer. There are 18 closely related species of Malassezia and all have an unusually small amount of genetic material compared with other types of yeast. In yeast, like in humans, the genetic material is divided among several chromosomes. The number of chromosomes in different Malassezia species varies between six and nine. A region of each chromosome known as the centromere is responsible for ensuring that the equal numbers of chromosomes are passed on to their offspring. This means that any defects in centromeres can lead to the daughter yeast cells inheriting unequal numbers of chromosomes. Changes in chromosome number can drive the evolution of new species, but it remains unclear if and how centromere loss may have contributed to the evolution of Malassezia species. Sankaranarayanan et al. have now used biochemical, molecular genetic, and comparative genomic approaches to study the chromosomes of Malassezia species. The experiments revealed that nine Malassezia species had centromeres that shared common features such as being rich in adenine and thymine nucleotides, two of the building blocks of DNA. Sankaranarayanan et al. propose that these adenines and thymines make the centromeres more fragile leading to occasional breaks. This may have contributed to the loss of centromeres in some Malassezia cells and helped new species to evolve with fewer chromosomes. A better understanding of how Malassezia organize their genetic material should enable in-depth studies of how these yeasts interact with their human hosts and how they contribute to skin disease, cancer, Crohn’s disease and other health conditions. More broadly, these findings may help scientists to better understand how changes in chromosomes cause new species to evolve.
© 2020, Sankaranarayanan et al.
Conflict of interest statement
SS, GI, MC, MR, BT, PG, RV, SS, RS, CT, TD, JH, KS No competing interests declared
Figures

References
-
- Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, Shadaloey SA, Wu D, Preiss P, Verma N, Guo Y, Saxena A, Vardhan M, Diskin B, Wang W, Leinwand J, Kurz E, Kochen Rossi JA, Hundeyin M, Zambrinis C, Li X, Saxena D, Miller G. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature. 2019;574:264–267. doi: 10.1038/s41586-019-1608-2. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
