

Transcriptomic and epigenetic mechanisms underlying myeloid diversity in the lung
- PMID: 31959980
- PMCID: PMC7667722
- DOI: 10.1038/s41590-019-0582-z
Transcriptomic and epigenetic mechanisms underlying myeloid diversity in the lung
Abstract
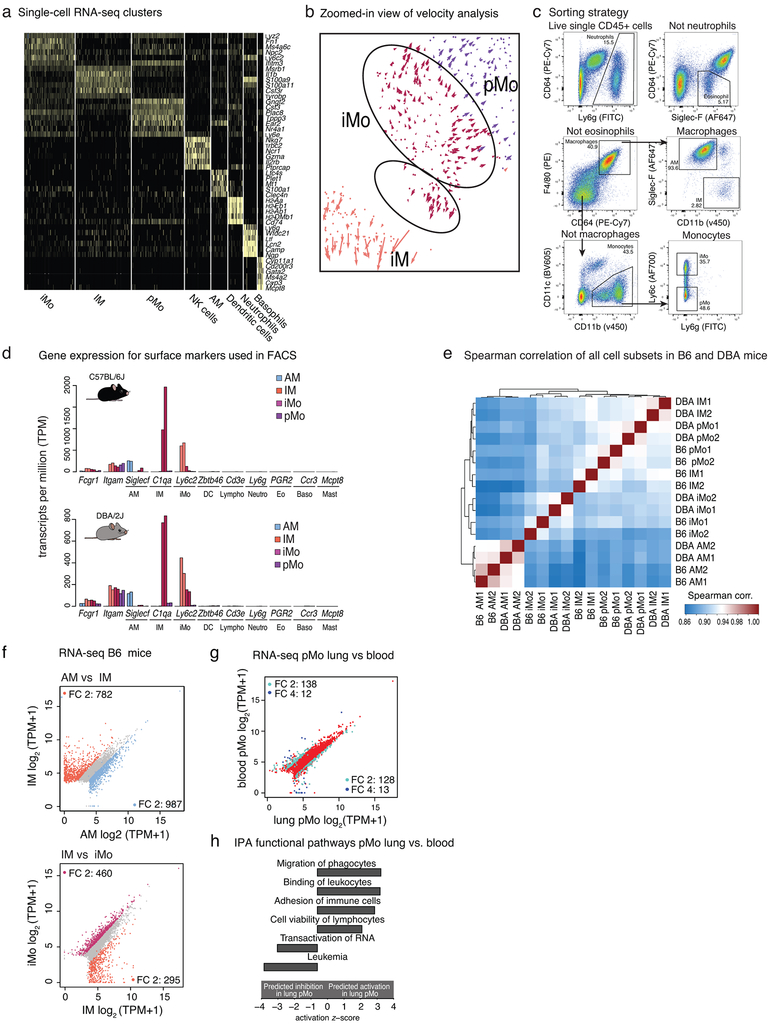
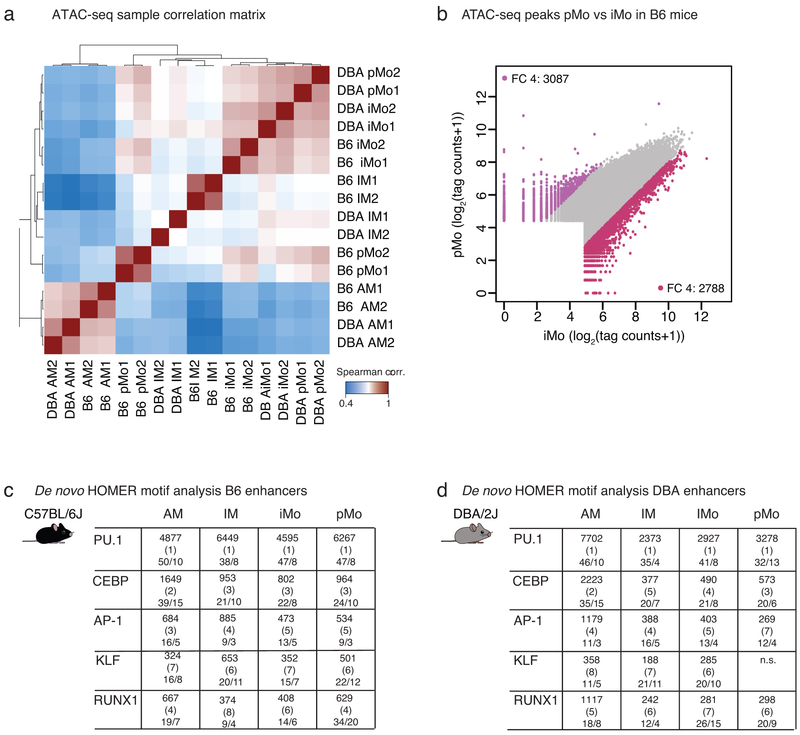
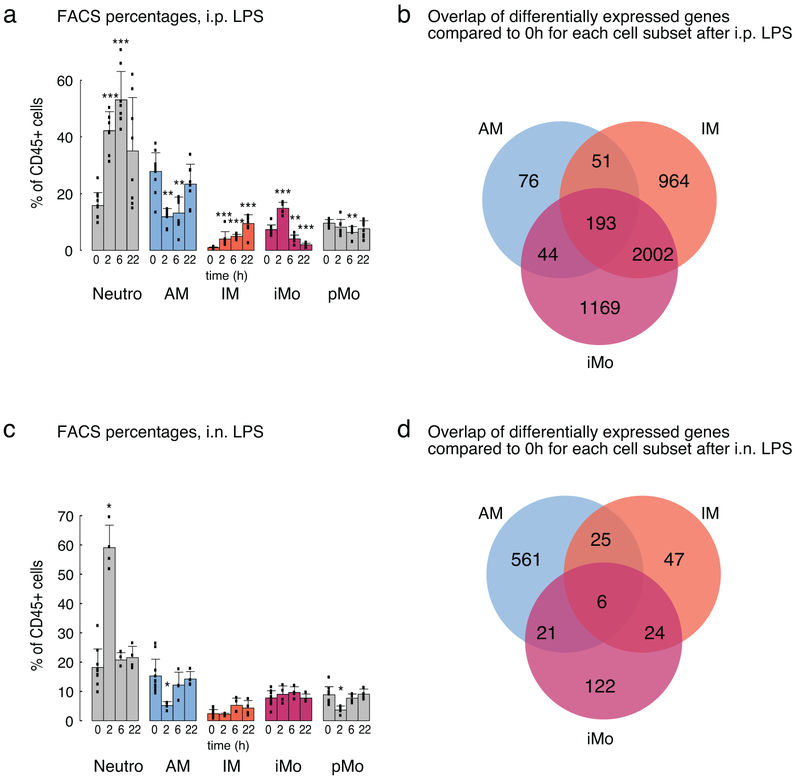
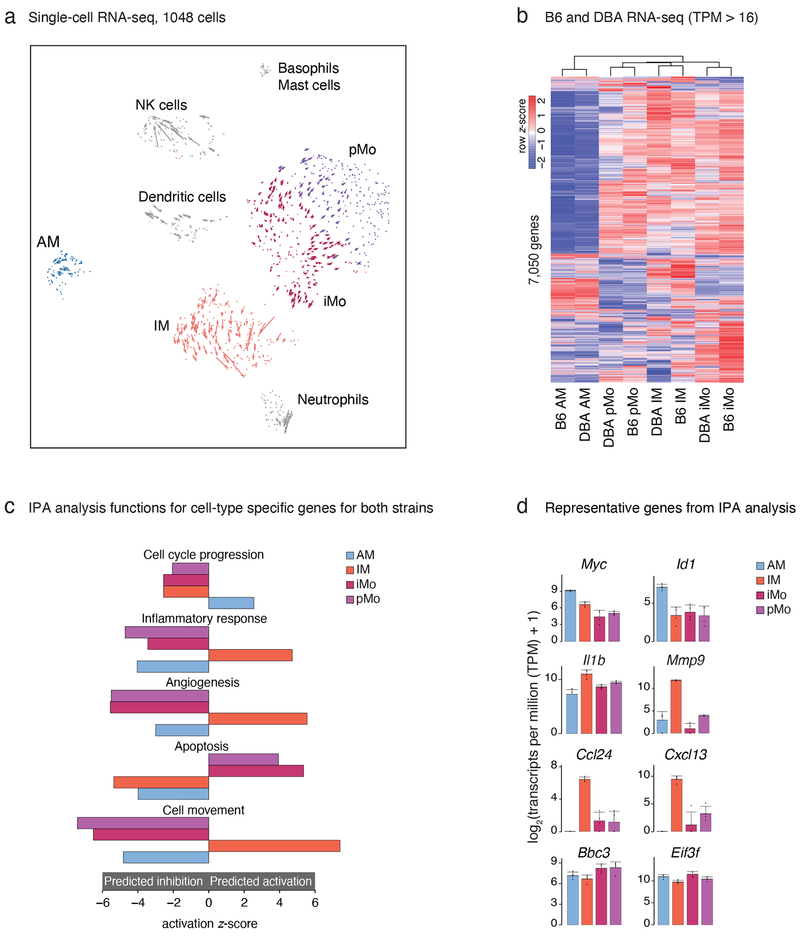
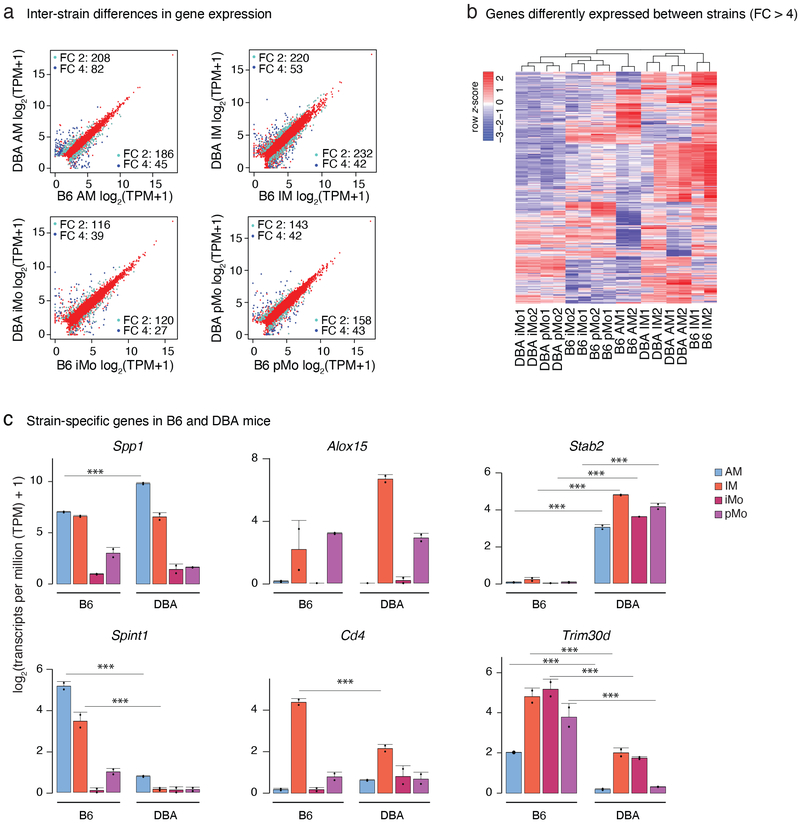
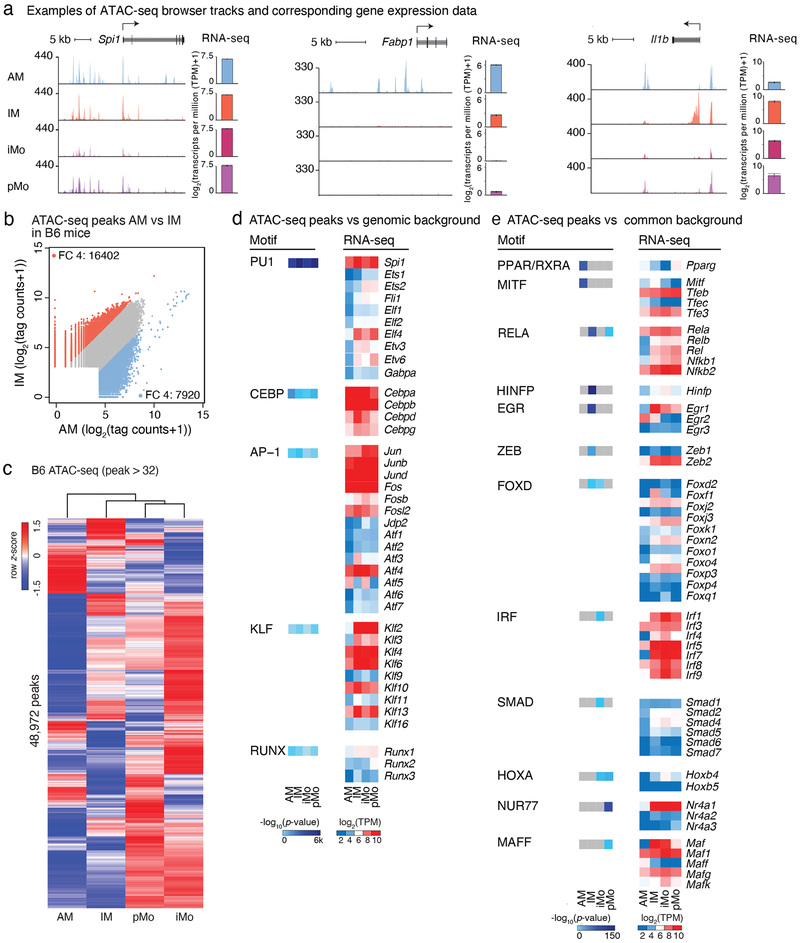
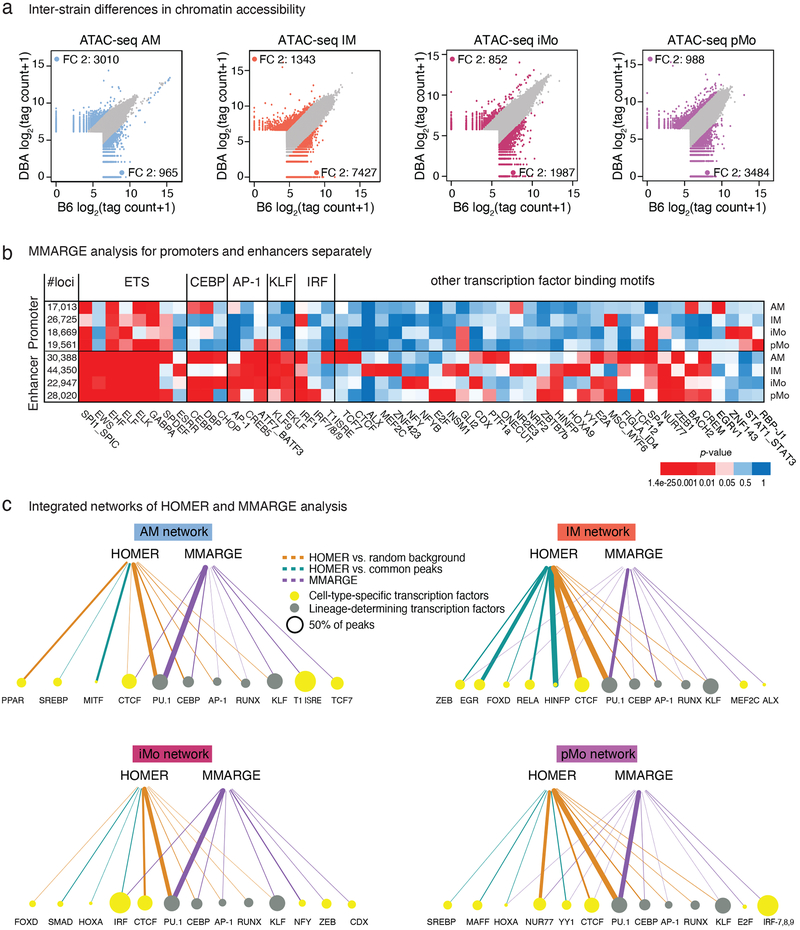
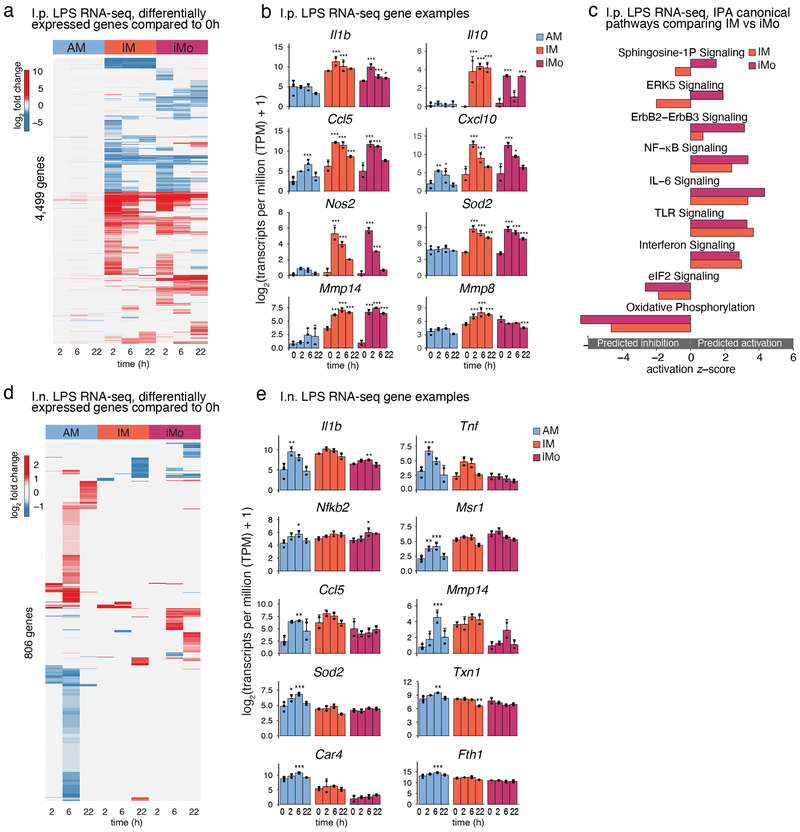
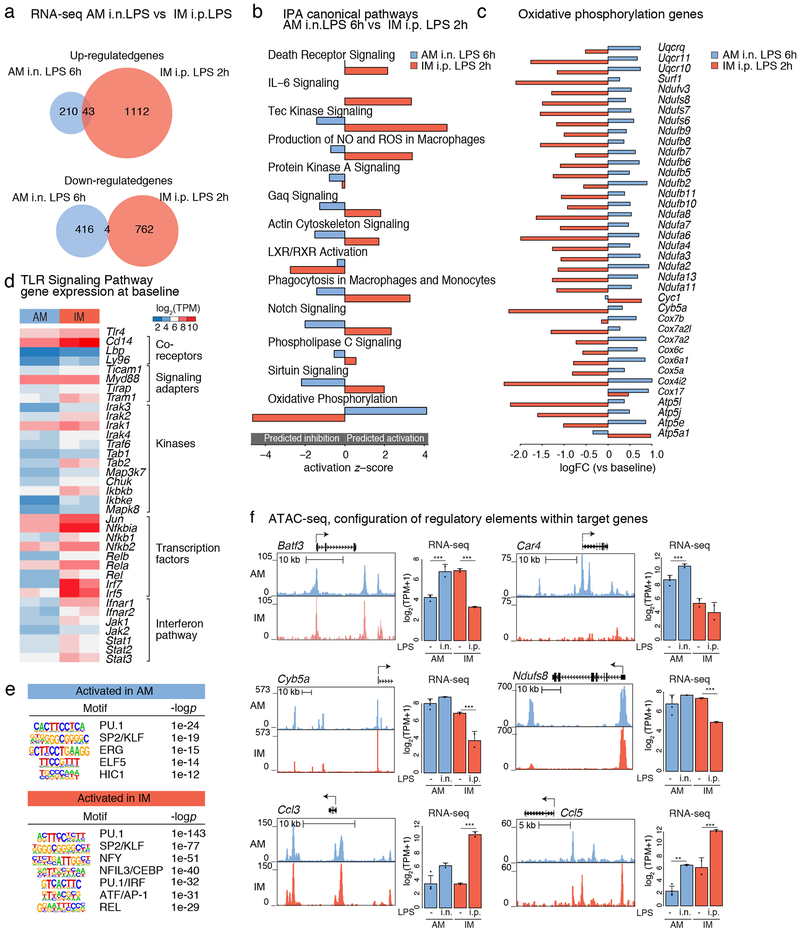
The lung is inhabited by resident alveolar and interstitial macrophages as well as monocytic cells that survey lung tissues. Each cell type plays distinct functional roles under homeostatic and inflammatory conditions, but mechanisms establishing their molecular identities and functional potential remain poorly understood. In the present study, systematic evaluation of transcriptomes and open chromatin of alveolar macrophages (AMs), interstitial macrophages (IMs) and lung monocytes from two mouse strains enabled inference of common and cell-specific transcriptional regulators. We provide evidence that these factors drive selection of regulatory landscapes that specify distinct phenotypes of AMs and IMs and entrain qualitatively different responses to toll-like receptor 4 signaling in vivo. These studies reveal a striking divergence in a fundamental innate immune response pathway in AMs and establish a framework for further understanding macrophage diversity in the lung.
Conflict of interest statement
Declaration of interests
The authors declare no competing interests.
Figures
Comment in
-
Diversity at the border.Nat Immunol. 2020 Feb;21(2):112-114. doi: 10.1038/s41590-019-0585-9. Nat Immunol. 2020. PMID: 31959978 No abstract available.
References
References (Methods only)
-
- Picelli S et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10, 1096–1098 (2013). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
