

Genetic studies of urinary metabolites illuminate mechanisms of detoxification and excretion in humans
- PMID: 31959995
- PMCID: PMC7484970
- DOI: 10.1038/s41588-019-0567-8
Genetic studies of urinary metabolites illuminate mechanisms of detoxification and excretion in humans
Abstract
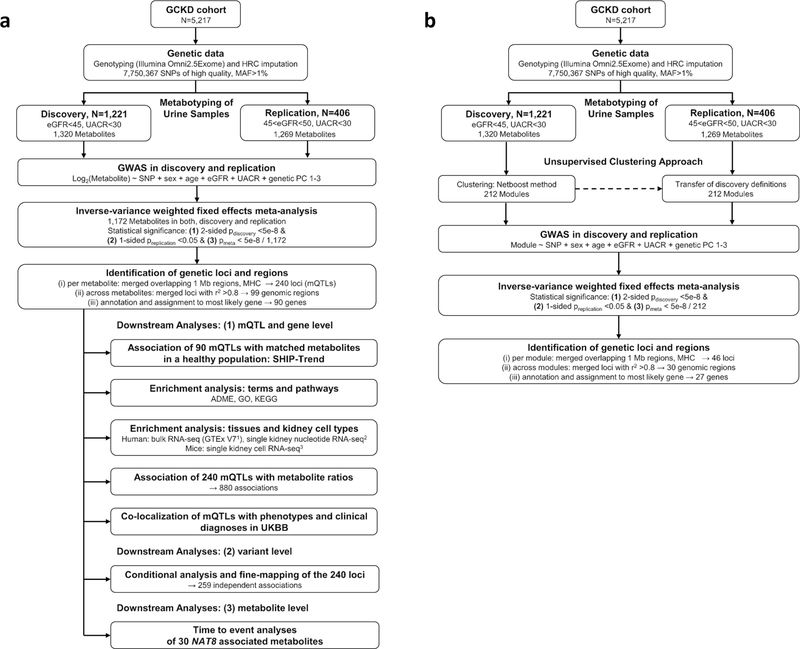
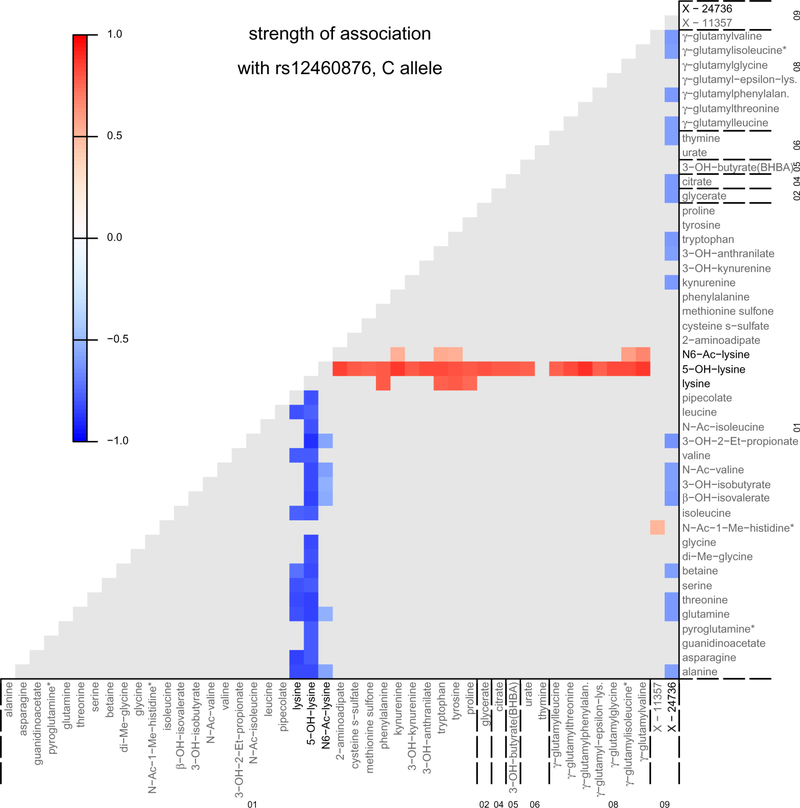
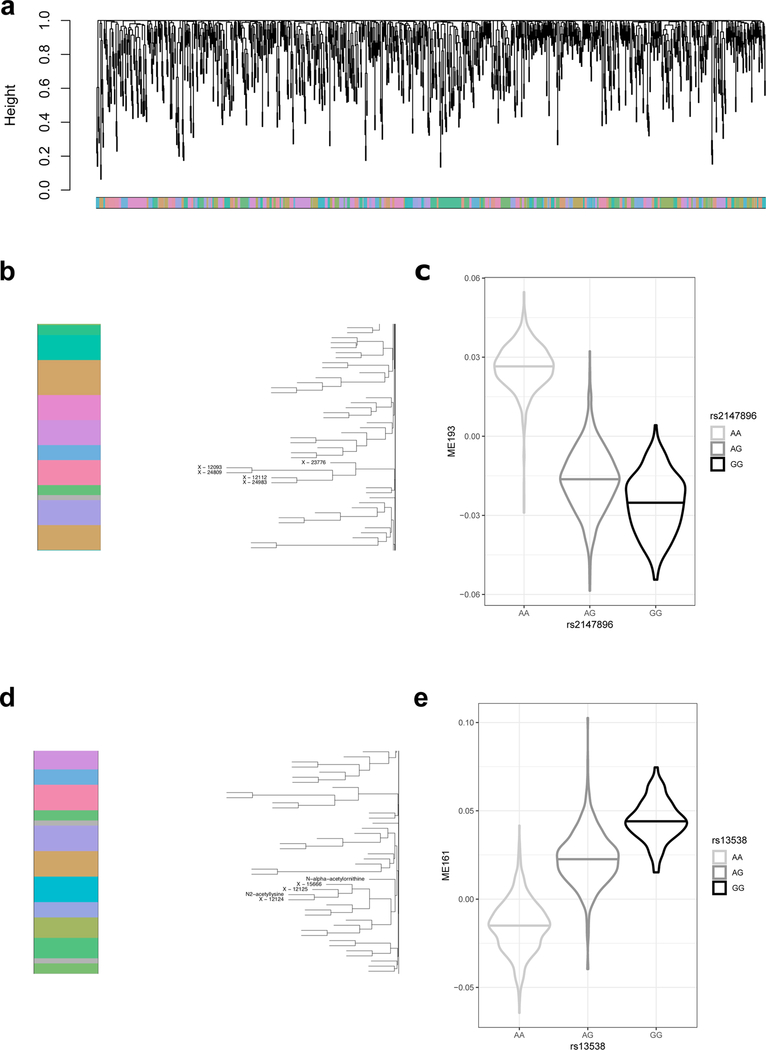
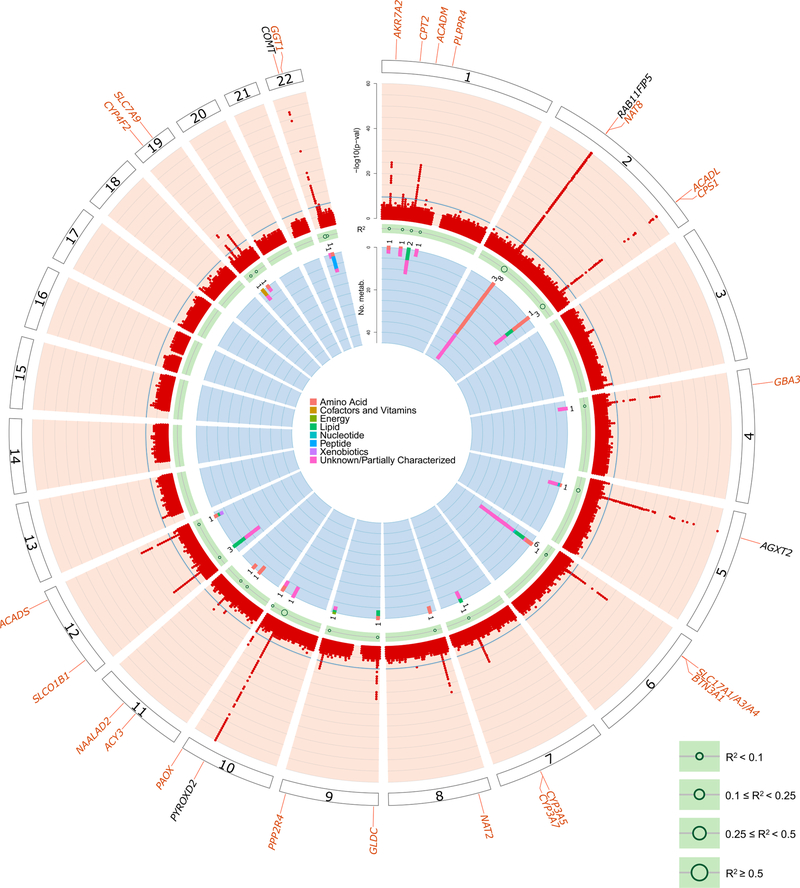
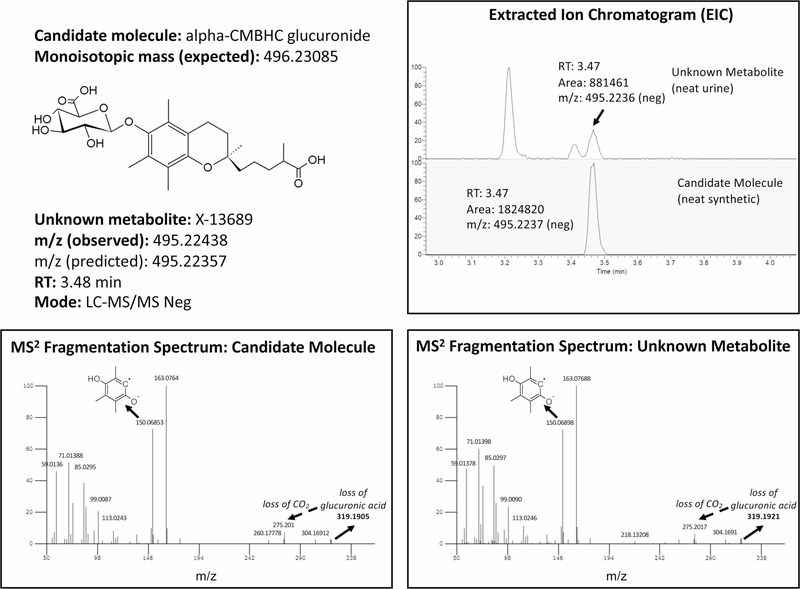
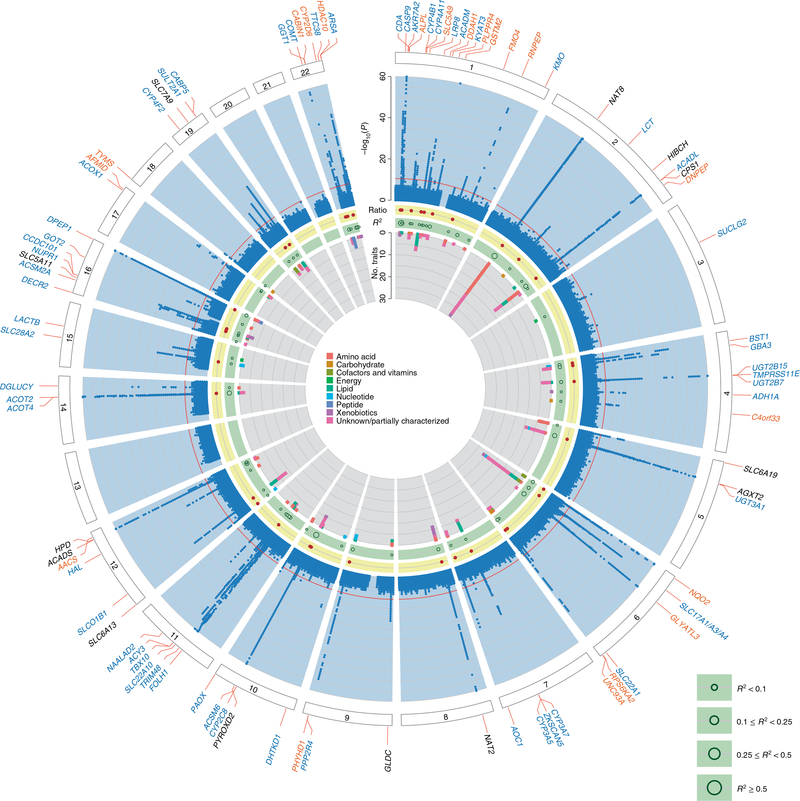
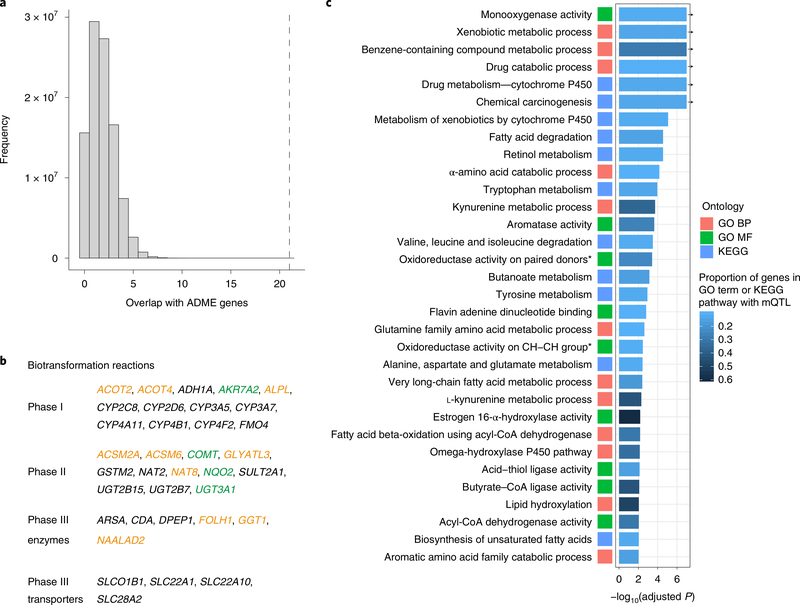
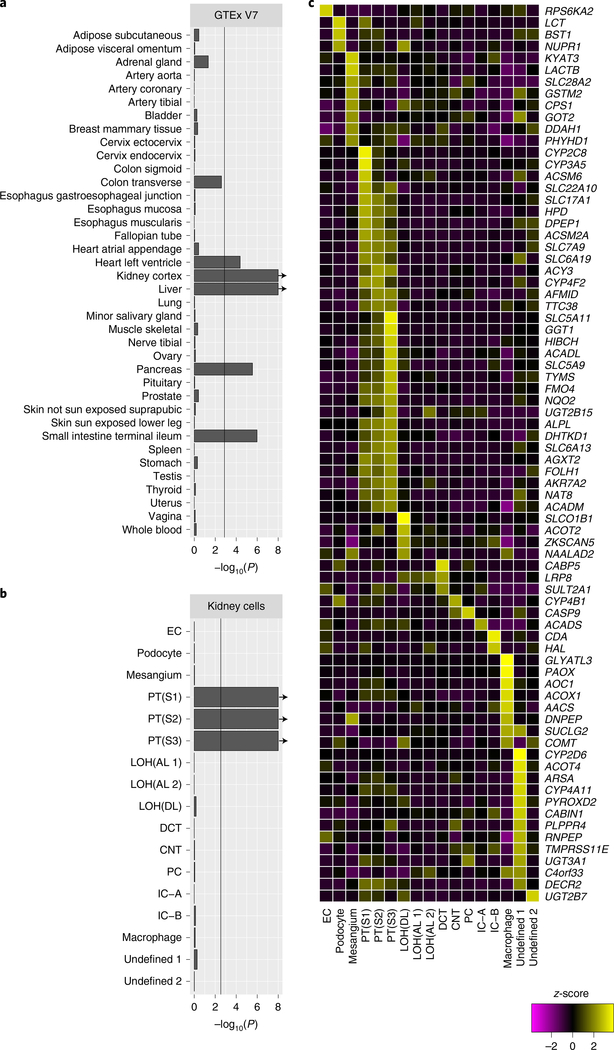
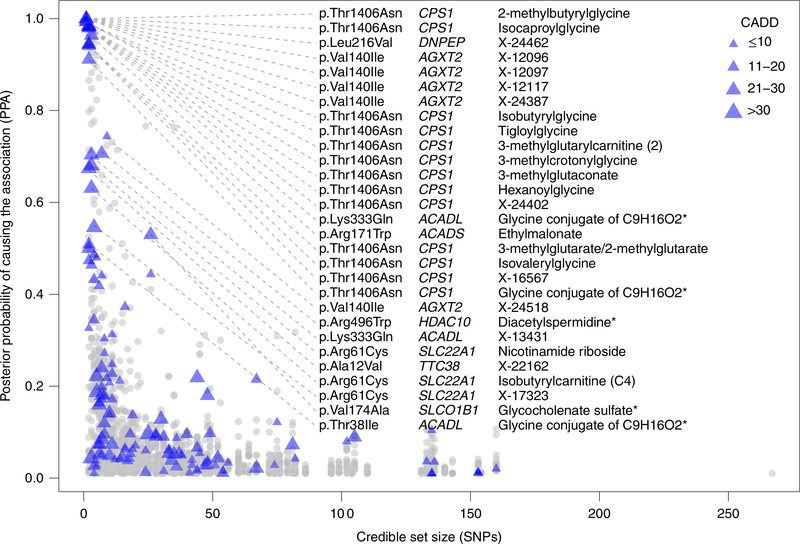
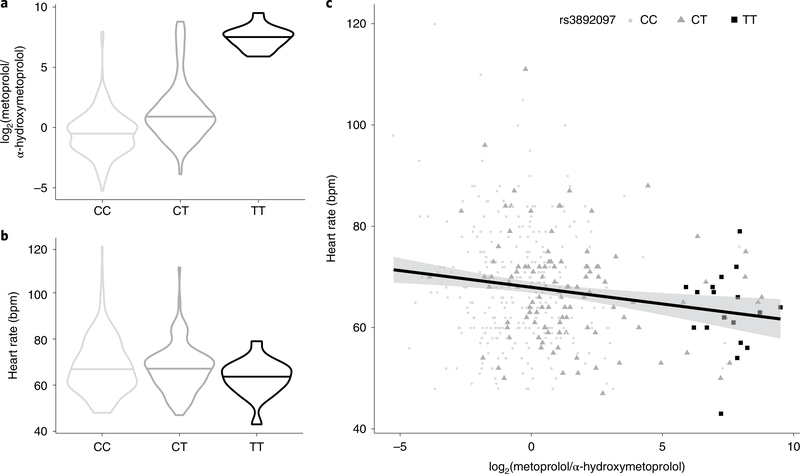
The kidneys integrate information from continuous systemic processes related to the absorption, distribution, metabolism and excretion (ADME) of metabolites. To identify underlying molecular mechanisms, we performed genome-wide association studies of the urinary concentrations of 1,172 metabolites among 1,627 patients with reduced kidney function. The 240 unique metabolite-locus associations (metabolite quantitative trait loci, mQTLs) that were identified and replicated highlight novel candidate substrates for transport proteins. The identified genes are enriched in ADME-relevant tissues and cell types, and they reveal novel candidates for biotransformation and detoxification reactions. Fine mapping of mQTLs and integration with single-cell gene expression permitted the prioritization of causal genes, functional variants and target cell types. The combination of mQTLs with genetic and health information from 450,000 UK Biobank participants illuminated metabolic mediators, and hence, novel urinary biomarkers of disease risk. This comprehensive resource of genetic targets and their substrates is informative for ADME processes in humans and is relevant to basic science, clinical medicine and pharmaceutical research.
Conflict of interest statement
Competing interests
R.P.M. is an employee of Metabolon and, as such, has affiliations with or financial involvement with Metabolon. All other authors have no competing interests.
Figures




Comment in
-
mGWAS: next generation genetic prediction in kidney disease.Nat Rev Nephrol. 2020 May;16(5):255-256. doi: 10.1038/s41581-020-0270-0. Nat Rev Nephrol. 2020. PMID: 32231316 Free PMC article.
References
-
- Caldwell J, Gardner I & Swales N An introduction to drug disposition: the basic principles of absorption, distribution, metabolism, and excretion. Toxicol. Pathol 23, 102–114 (1995). - PubMed
-
- Köttgen A, Raffler J, Sekula P & Kastenmuller G Genome-wide association studies of metabolite concentrations (mGWAS): Relevance for nephrology. Semin. Nephrol 38, 151–174 (2018). - PubMed
-
- Homuth G, Teumer A, Volker U & Nauck M A description of large-scale metabolomics studies: increasing value by combining metabolomics with genome-wide SNP genotyping and transcriptional profiling. J. Endocrinol. 215, 17–28 (2012). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
