

Ischemia and Reperfusion Injury in Kidney Transplantation: Relevant Mechanisms in Injury and Repair
- PMID: 31963521
- PMCID: PMC7019324
- DOI: 10.3390/jcm9010253
Ischemia and Reperfusion Injury in Kidney Transplantation: Relevant Mechanisms in Injury and Repair
Abstract
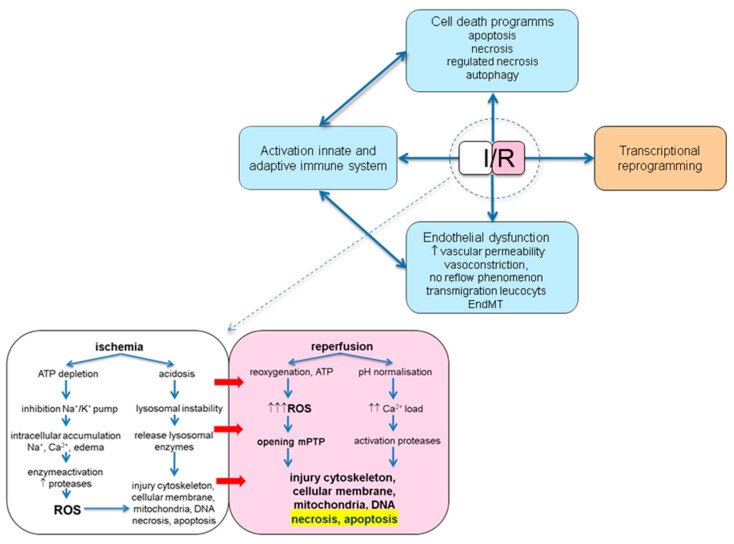
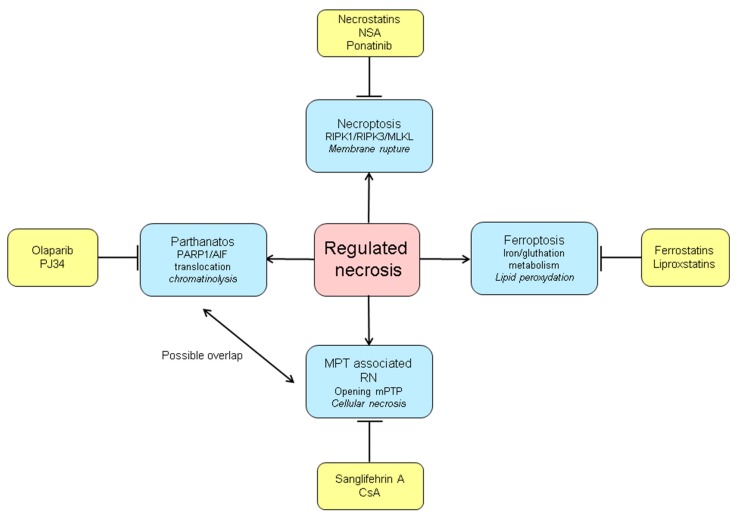
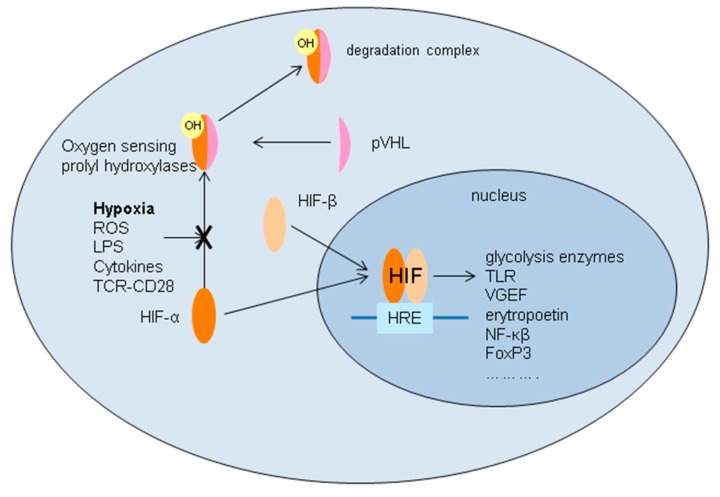
Ischemia and reperfusion injury (IRI) is a complex pathophysiological phenomenon, inevitable in kidney transplantation and one of the most important mechanisms for non- or delayed function immediately after transplantation. Long term, it is associated with acute rejection and chronic graft dysfunction due to interstitial fibrosis and tubular atrophy. Recently, more insight has been gained in the underlying molecular pathways and signalling cascades involved, which opens the door to new therapeutic opportunities aiming to reduce IRI and improve graft survival. This review systemically discusses the specific molecular pathways involved in the pathophysiology of IRI and highlights new therapeutic strategies targeting these pathways.
Keywords: adaptive immune system; apoptosis; delayed graft function; endothelial dysfunction; hypoxic inducible factor; innate immune system; ischemia reperfusion injury; kidney transplantation; necrosis.
Conflict of interest statement
The authors declare no conflict of interest. Parts of this review are adapted from chapter 2 of the PhD thesis: Perioperative renal protective strategies in kidney transplantation, Gertrude J. Nieuwenhuijs-Moeke, 2019.
Figures






References
-
- World Health Organisation . Disease Burden and Mortality Estimates. WTO; Geneva, Switzerland: 2015. [(accessed on 15 January 2020)]. Available online: https://www.who.int/healthinfo/global_burden_disease/estimates/en/index1....
-
- Global Observatory on Donation and Transplantation. [(accessed on 15 January 2020)];2019 Available online: http://www.transplant-observatory.org.
-
- Cooper J.T., Chin L.T., Krieger N.R., Fernandez L.A., Foley D.P., Becker Y.T., Odorico J.S., Knechtle S.J., Kalayoglu M., Sollinger H.W., et al. Donation after cardiac death: The university of Wisconsin experience with renal transplantation. Am. J. Transplant. 2004;4:1490–1494. doi: 10.1111/j.1600-6143.2004.00531.x. - DOI - PubMed
-
- Koffman G., Gambaro G. Renal transplantation from non-heart-beating donors: A review of the European experience. J. Nephrol. 2003;16:334–341. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
