

Prevention of DNA Replication Stress by CHK1 Leads to Chemoresistance Despite a DNA Repair Defect in Homologous Recombination in Breast Cancer
- PMID: 31963582
- PMCID: PMC7017274
- DOI: 10.3390/cells9010238
Prevention of DNA Replication Stress by CHK1 Leads to Chemoresistance Despite a DNA Repair Defect in Homologous Recombination in Breast Cancer
Abstract
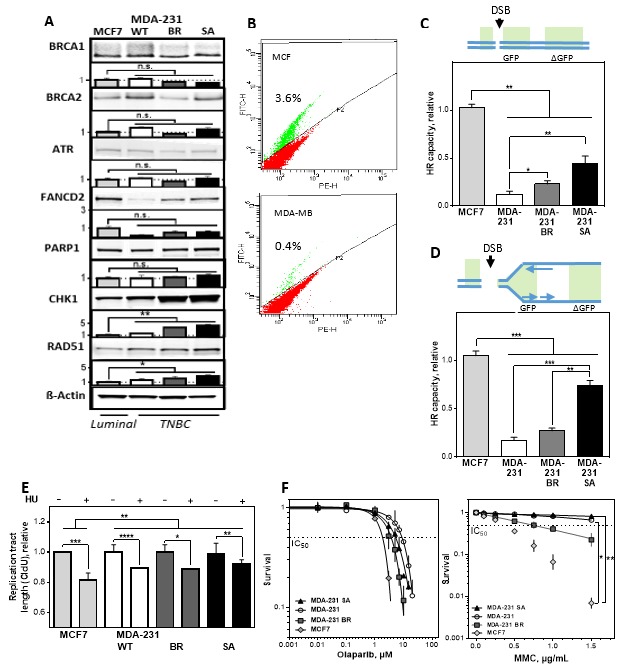
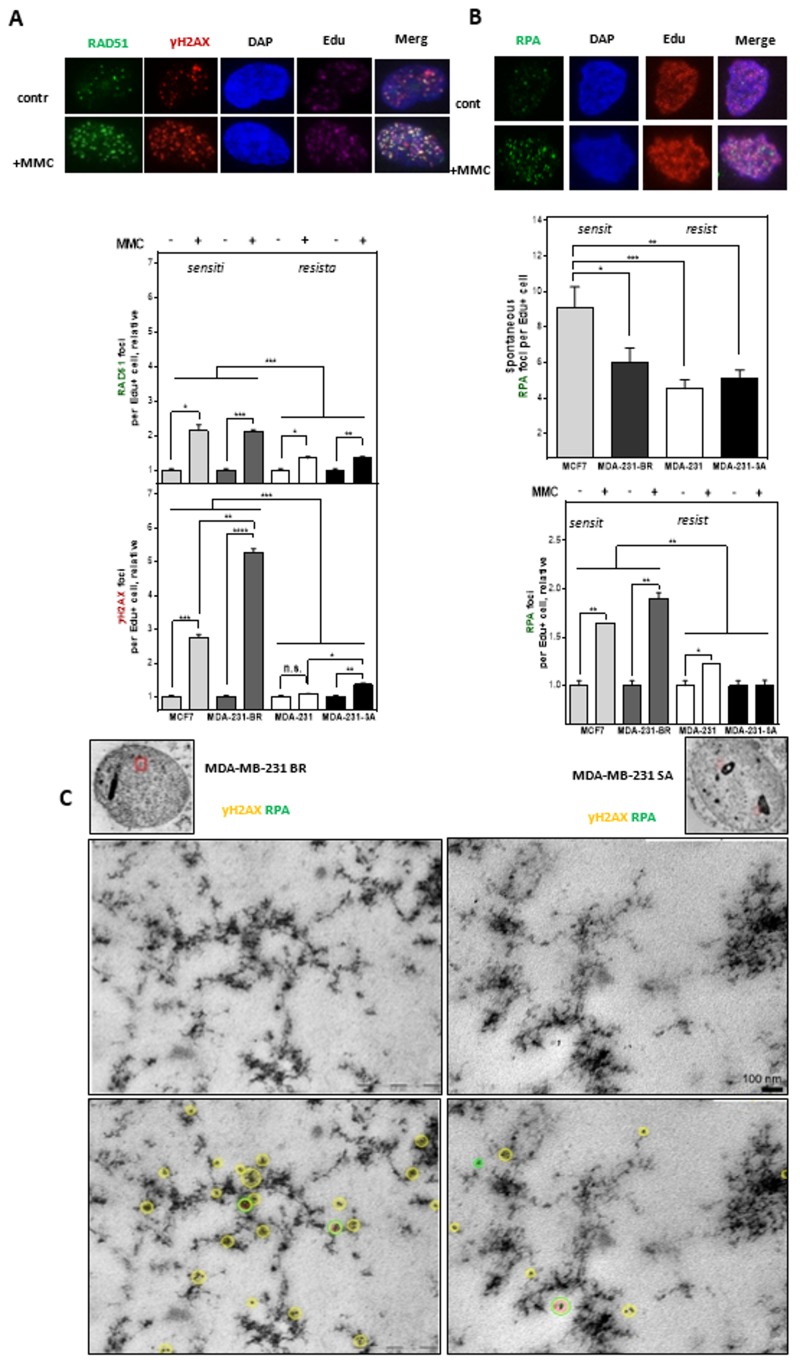
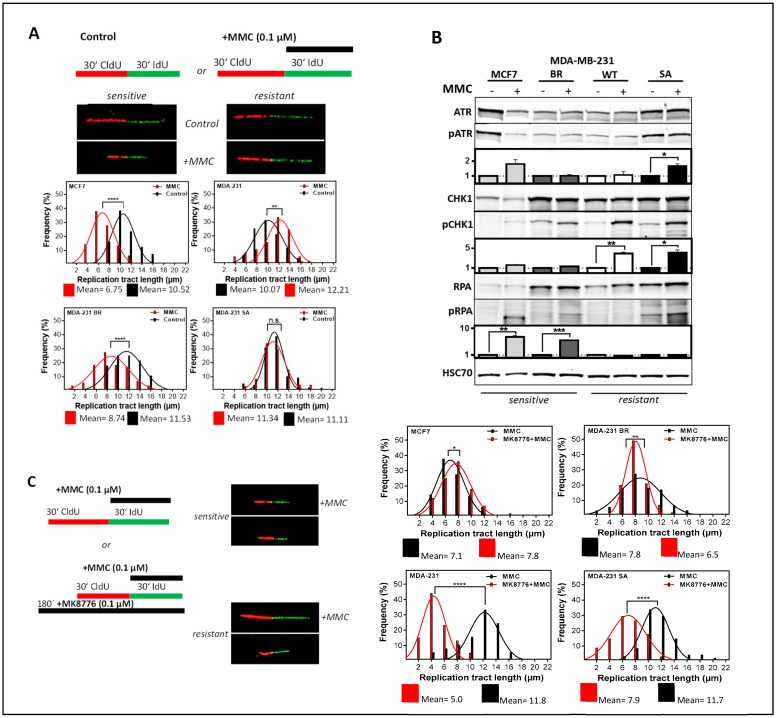
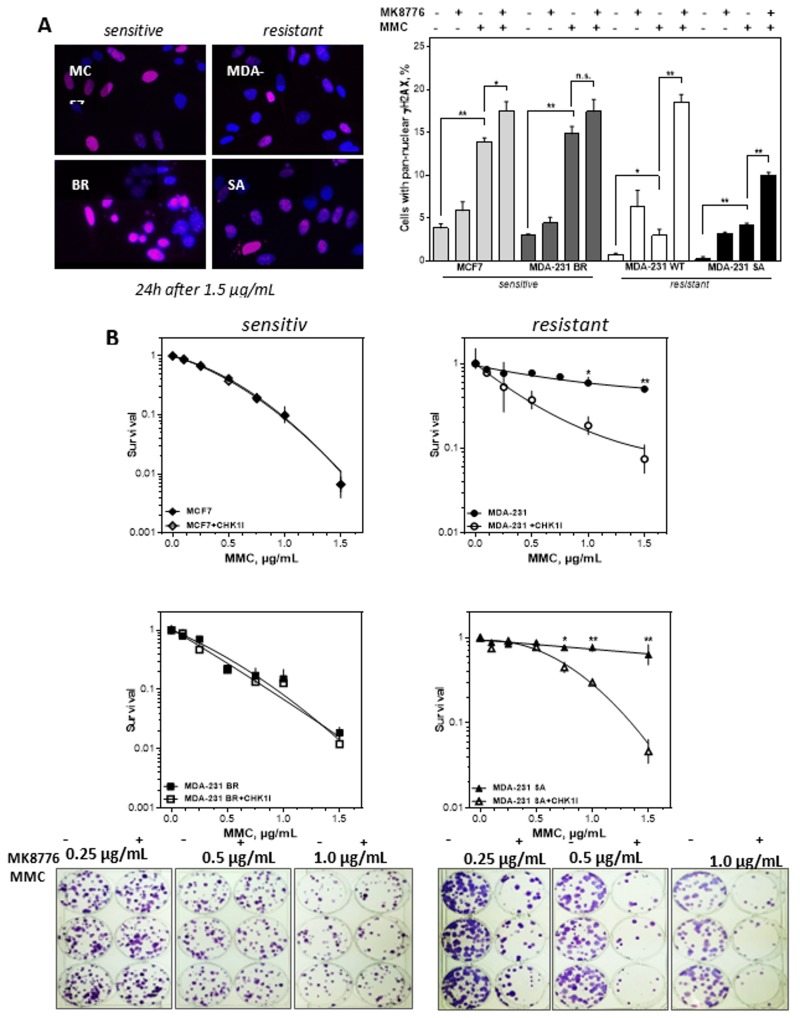
Chromosomal instability not only has a negative effect on survival in triple-negative breast cancer, but also on the well treatable subgroup of luminal A tumors. This suggests a general mechanism independent of subtypes. Increased chromosomal instability (CIN) in triple-negative breast cancer (TNBC) is attributed to a defect in the DNA repair pathway homologous recombination. Homologous recombination (HR) prevents genomic instability by repair and protection of replication. It is unclear whether genetic alterations actually lead to a repair defect or whether superior signaling pathways are of greater importance. Previous studies focused exclusively on the repair function of HR. Here, we show that the regulation of HR by the intra-S-phase damage response at the replication is of overriding importance. A damage response activated by Ataxia telangiectasia and Rad3 related-checkpoint kinase 1 (ATR-CHK1) can prevent replication stress and leads to resistance formation. CHK1 thus has a preferred role over HR in preventing replication stress in TNBC. The signaling cascade ATR-CHK1 can compensate for a double-strand break repair error and lead to resistance of HR-deficient tumors. Established methods for the identification of HR-deficient tumors for Poly(ADP-Ribose)-Polymerase 1 (PARP1) inhibitor therapies should be extended to include analysis of candidates for intra-S phase damage response.
Keywords: CHK1; CIN70 score; DNA-damage response (DDR); chromosomal instability (CIN); homologous recombination (HR); triple-negative breast cancer (TNBC).
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

References
-
- Birkbak N.J., Eklund A.C., Li Q., McClelland S.E., Endesfelder D., Tan P., Tan I.B., Richardson A.L., Szallasi Z., Swanton C. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011;71:3447–3452. doi: 10.1158/0008-5472.CAN-10-3667. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
