

Two integrated and highly predictive functional analysis-based procedures for the classification of MSH6 variants in Lynch syndrome
- PMID: 31965077
- PMCID: PMC7200593
- DOI: 10.1038/s41436-019-0736-2
Two integrated and highly predictive functional analysis-based procedures for the classification of MSH6 variants in Lynch syndrome
Abstract
Purpose: Variants in the DNA mismatch repair (MMR) gene MSH6, identified in individuals suspected of Lynch syndrome, are difficult to classify owing to the low cancer penetrance of defects in that gene. This not only obfuscates personalized health care but also the development of a rapid and reliable classification procedure that does not require clinical data.
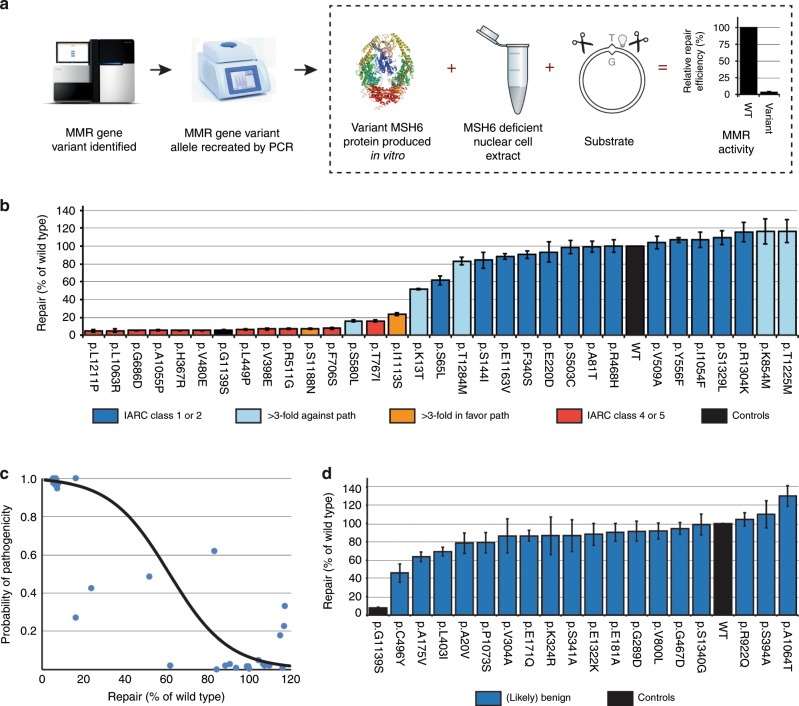
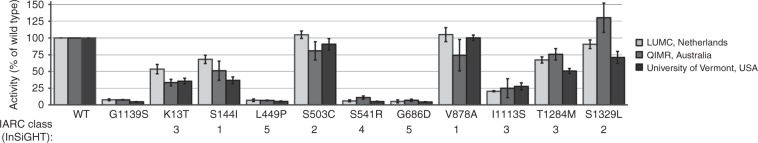
Methods: The complete in vitro MMR activity (CIMRA) assay was calibrated against clinically classified MSH6 variants and, employing Bayes' rule, integrated with computational predictions of pathogenicity. To enable the validation of this two-component classification procedure we have employed a genetic screen to generate a large set of inactivating Msh6 variants, as proxies for pathogenic variants.
Results: The genetic screen-derived variants established that the two-component classification procedure displays high sensitivities and specificities. Moreover, these inactivating variants enabled the direct reclassification of human variants of uncertain significance (VUS) as (likely) pathogenic.
Conclusion: The two-component classification procedure and the genetic screens provide complementary approaches to rapidly and cost-effectively classify the large majority of human MSH6 variants. The approach followed here provides a template for the classification of variants in other disease-predisposing genes, facilitating the translation of personalized genomics into personalized health care.
Keywords: DNA mismatch repair; Lynch syndrome; MSH6; functional analysis-based classification; variants of uncertain significance.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures


References
-
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
