

Trypanosoma brucei ATR Links DNA Damage Signaling during Antigenic Variation with Regulation of RNA Polymerase I-Transcribed Surface Antigens
- PMID: 31968257
- PMCID: PMC6988115
- DOI: 10.1016/j.celrep.2019.12.049
Trypanosoma brucei ATR Links DNA Damage Signaling during Antigenic Variation with Regulation of RNA Polymerase I-Transcribed Surface Antigens
Abstract
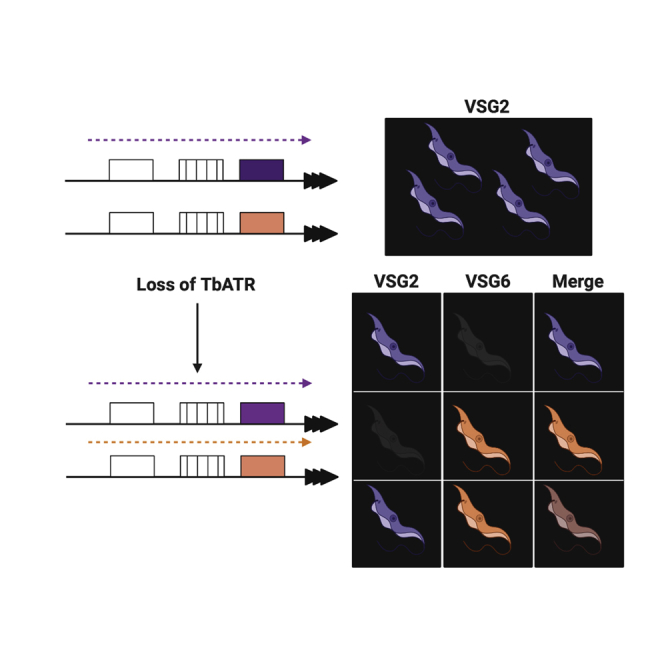
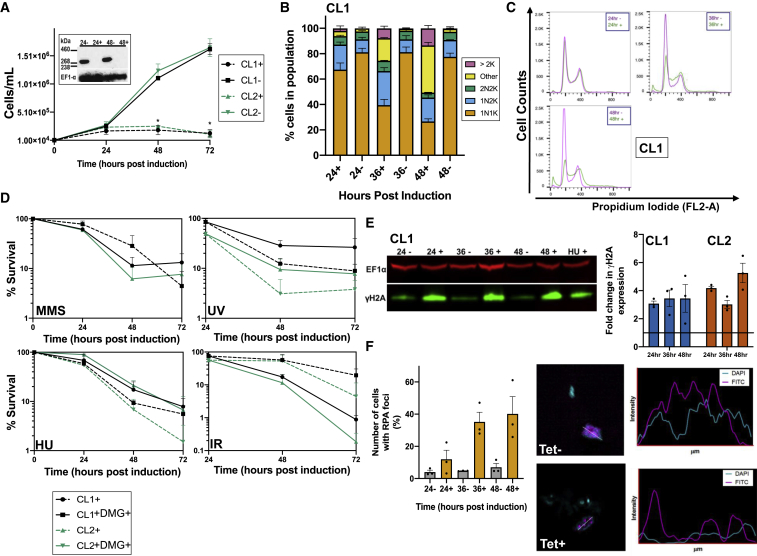
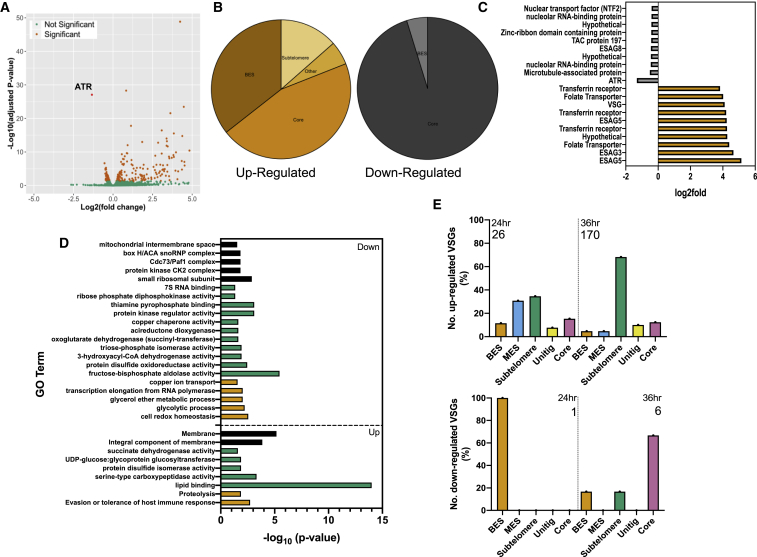
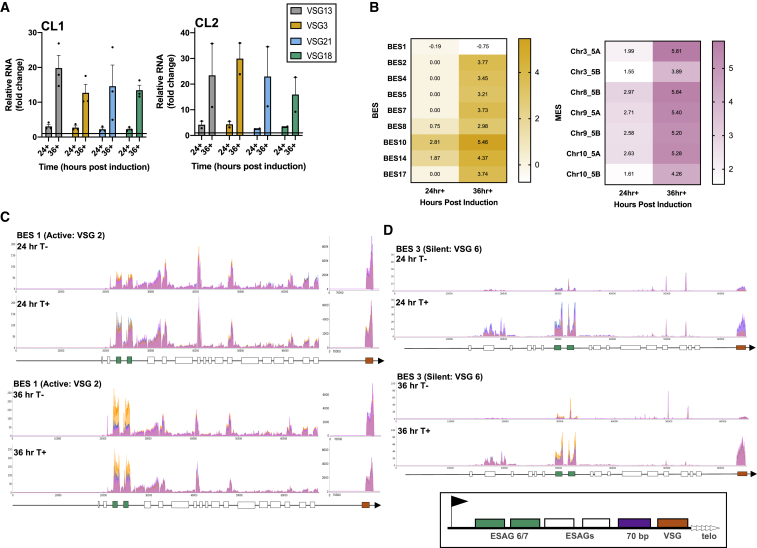
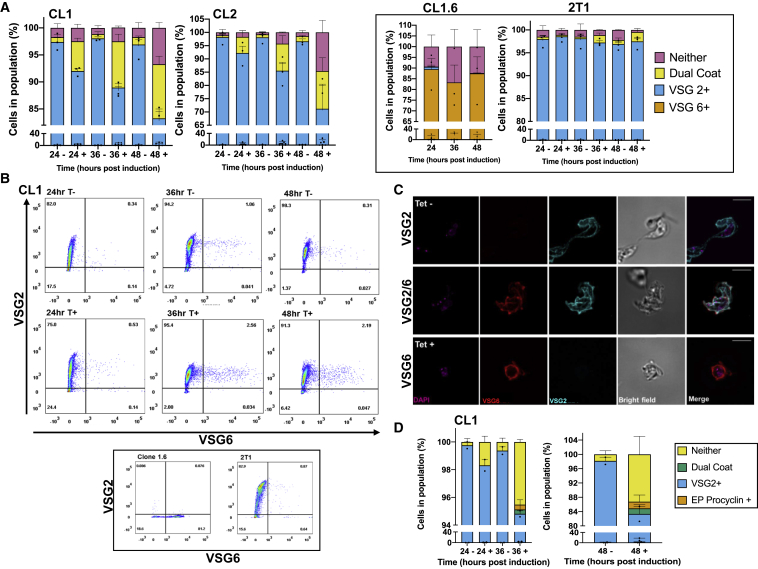
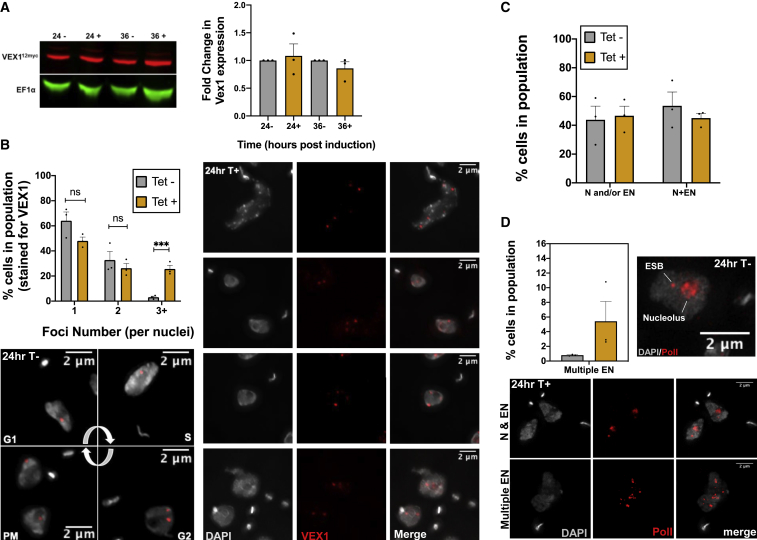
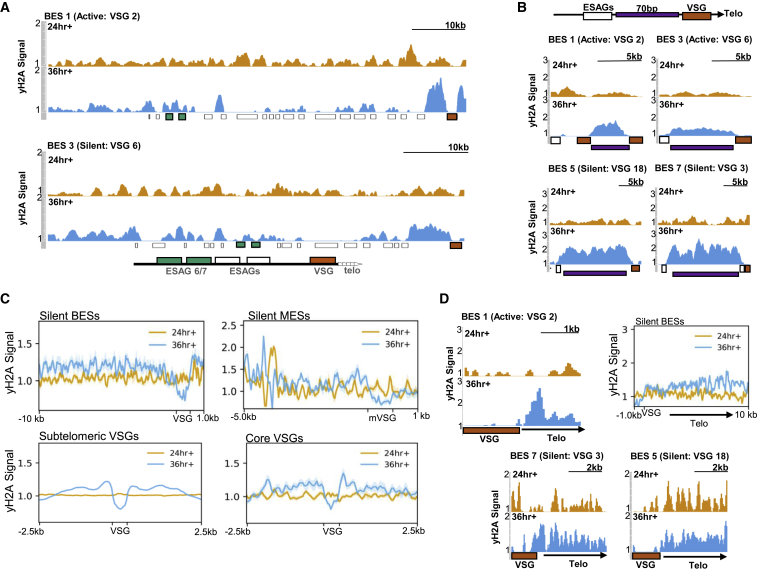
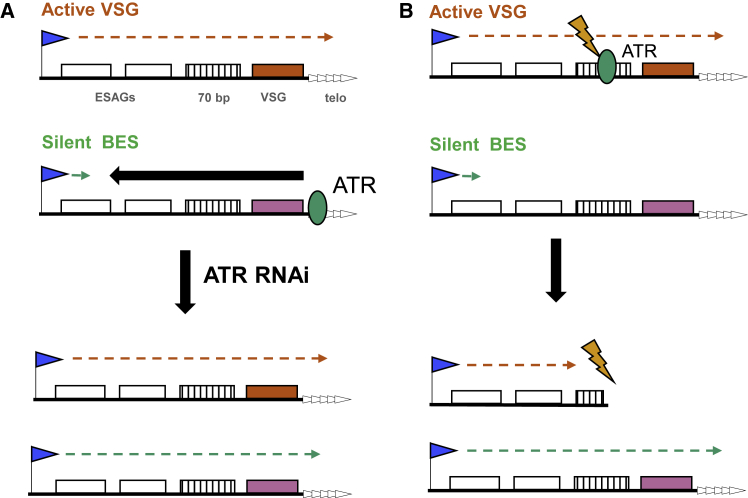
Trypanosoma brucei evades mammalian immunity by using recombination to switch its surface-expressed variant surface glycoprotein (VSG), while ensuring that only one of many subtelomeric multigene VSG expression sites are transcribed at a time. DNA repair activities have been implicated in the catalysis of VSG switching by recombination, not transcriptional control. How VSG switching is signaled to guide the appropriate reaction or to integrate switching into parasite growth is unknown. Here, we show that the loss of ATR, a DNA damage-signaling protein kinase, is lethal, causing nuclear genome instability and increased VSG switching through VSG-localized damage. Furthermore, ATR loss leads to the increased transcription of silent VSG expression sites and expression of mixed VSGs on the cell surface, effects that are associated with the altered localization of RNA polymerase I and VEX1. This work shows that ATR acts in antigenic variation both through DNA damage signaling and surface antigen expression control.
Keywords: ATR; DNA damage signaling; DNA replication stress; RNA polymerase I; Trypanosoma brucei; antigenic variation; immune evasion; monoallelic expression; protein kinase; variant surface glycoprotein.
Copyright © 2019 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
References
-
- Alexa A., Rahnenfuhrer J., Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 104111/WT_/Wellcome Trust/United Kingdom
- BB/N016165/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/M028909/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/K006495/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Miscellaneous
