

Aberrantly High Levels of Somatic LINE-1 Expression and Retrotransposition in Human Neurological Disorders
- PMID: 31969897
- PMCID: PMC6960195
- DOI: 10.3389/fgene.2019.01244
Aberrantly High Levels of Somatic LINE-1 Expression and Retrotransposition in Human Neurological Disorders
Abstract
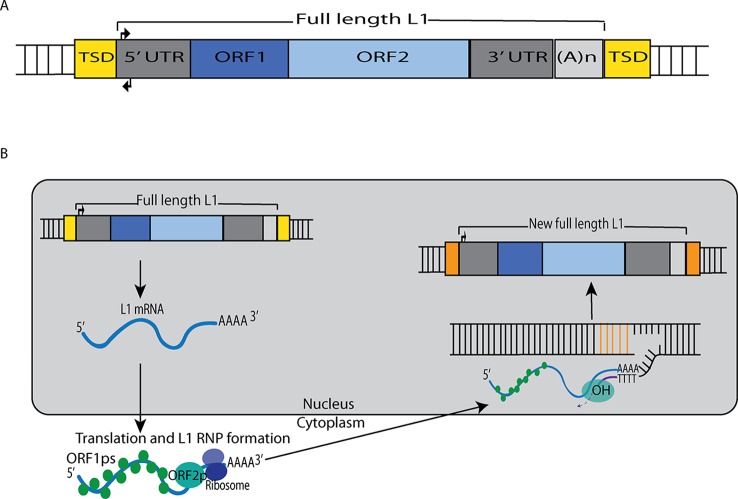
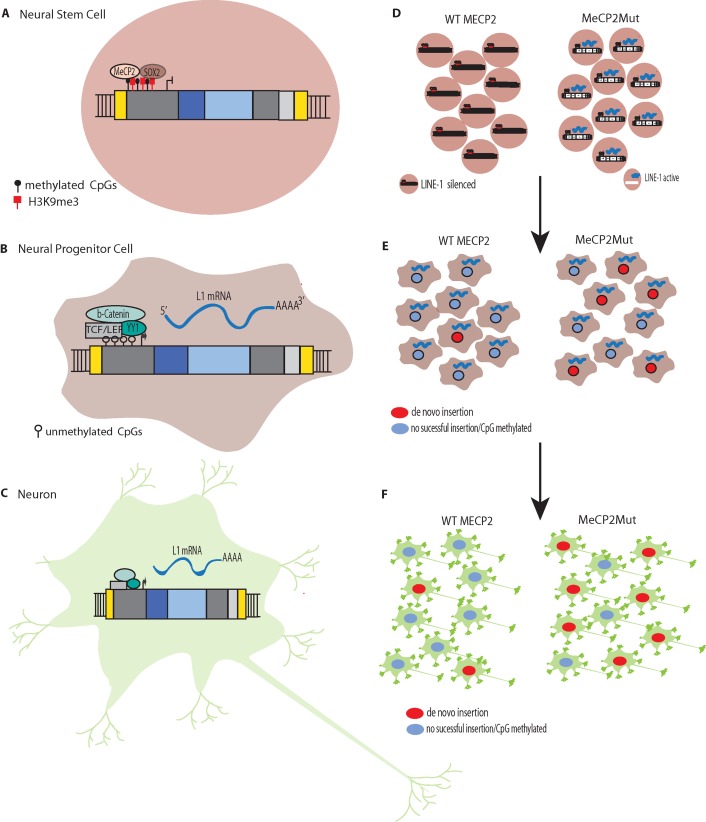
Retrotransposable elements (RTEs) have actively multiplied over the past 80 million years of primate evolution, and as a consequence, such elements collectively occupy ∼ 40% of the human genome. As RTE activity can have detrimental effects on the human genome and transcriptome, silencing mechanisms have evolved to restrict retrotransposition. The brain is the only known somatic tissue where RTEs are de-repressed throughout the life of a healthy human and each neuron in specific brain regions accumulates up to ∼13.7 new somatic L1 insertions (and perhaps more). However, even higher levels of somatic RTE expression and retrotransposition have been found in a number of human neurological disorders. This review is focused on how RTE expression and retrotransposition in neuronal tissues might contribute to the initiation and progression of these disorders. These disorders are discussed in three broad and sometimes overlapping categories: 1) disorders such as Rett syndrome, Aicardi-Goutières syndrome, and ataxia-telangiectasia, where expression/retrotransposition is increased due to mutations in genes that play a role in regulating RTEs in healthy cells, 2) disorders such as autism spectrum disorder, schizophrenia, and substance abuse disorders, which are thought to be caused by a combination of genetic and environmental stress factors, and 3) disorders associated with age, such as frontotemporal lobar degeneration (FTLD), amyotrophic lateral sclerosis (ALS), and normal aging, where there is a time-dependent accumulation of neurological degeneration, RTE copy number, and phenotypes. Research has revealed increased levels of RTE activity in many neurological disorders, but in most cases, a clear causal link between RTE activity and these disorders has not been well established. At the same time, even if increased RTE activity is a passenger and not a driver of disease, a detrimental effect is more likely than a beneficial one. Thus, a better understanding of the role of RTEs in neuronal tissues likely is an important part of understanding, preventing, and treating these disorders.
Keywords: L1; LINE; SINE; brain; neurological disease; retrotransposition; somatic.
Copyright © 2020 Terry and Devine.
Figures
References
-
- Baio J., Wiggins L., Christensen D. L., Maenner M. J., Daniels J., Warren Z., et al. (2018). Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 Sites, United States, 2014 . MMWR Surveill Summ. 67 (6), 1–23. 10.15585/mmwr.ss6706a1 - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous