

Current Status of AMOEBA-IL: A Multipolar/Polarizable Force Field for Ionic Liquids
- PMID: 31973103
- PMCID: PMC7037047
- DOI: 10.3390/ijms21030697
Current Status of AMOEBA-IL: A Multipolar/Polarizable Force Field for Ionic Liquids
Abstract
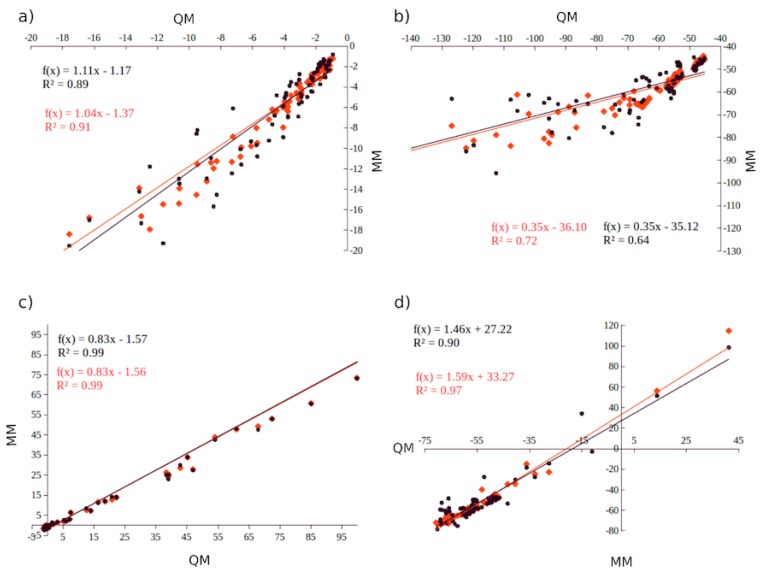
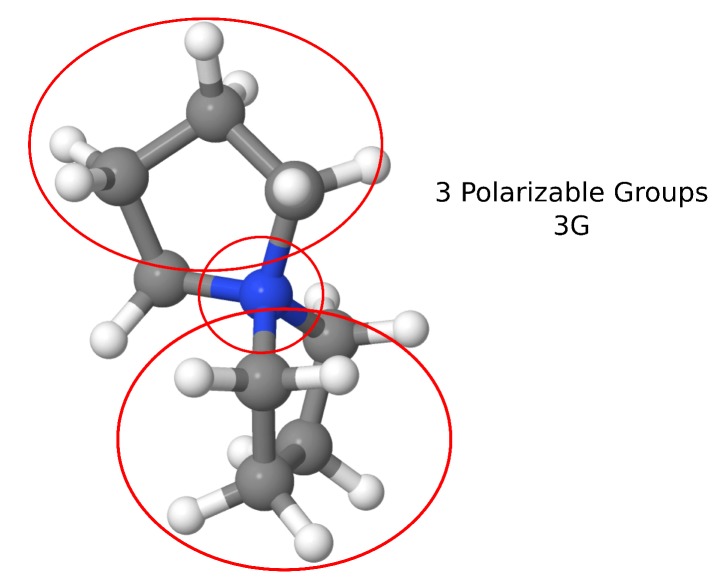
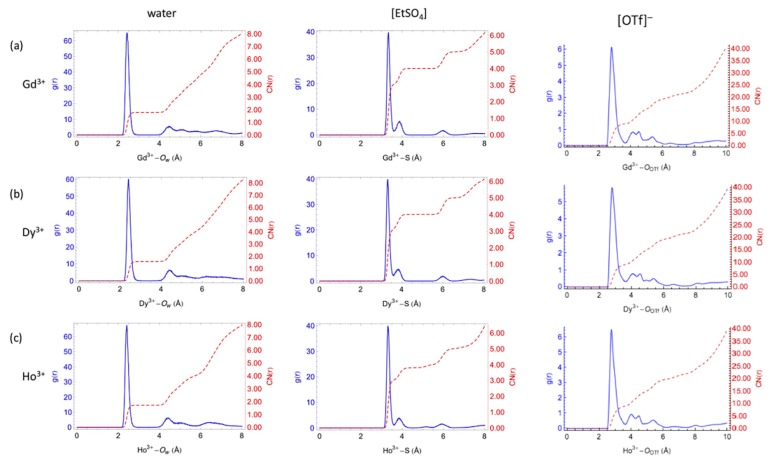
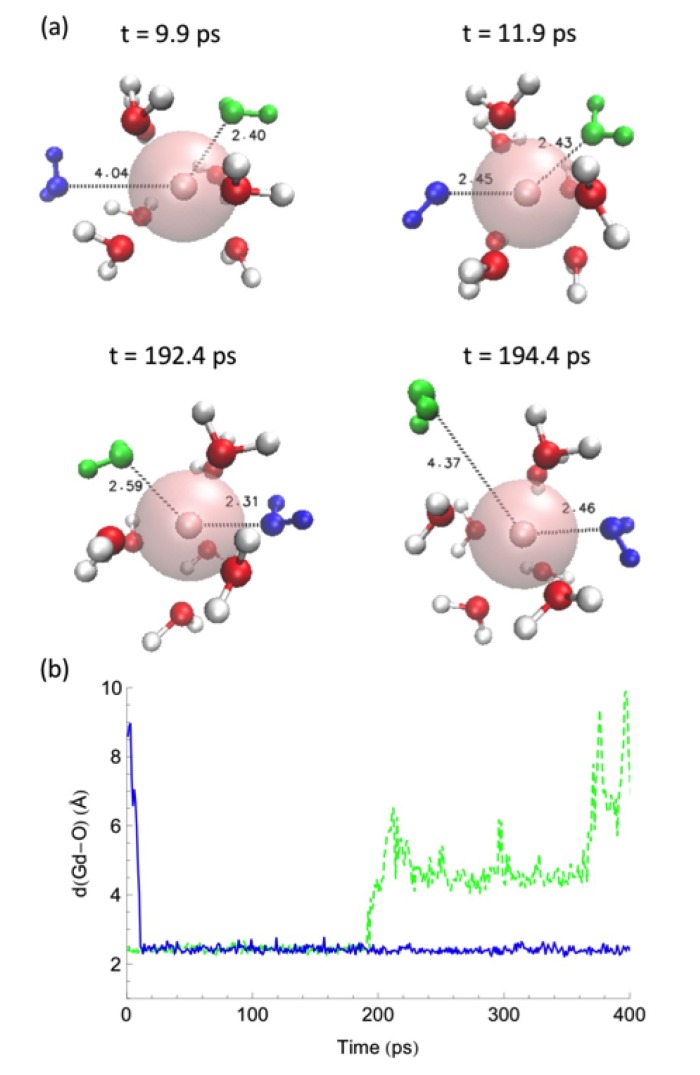
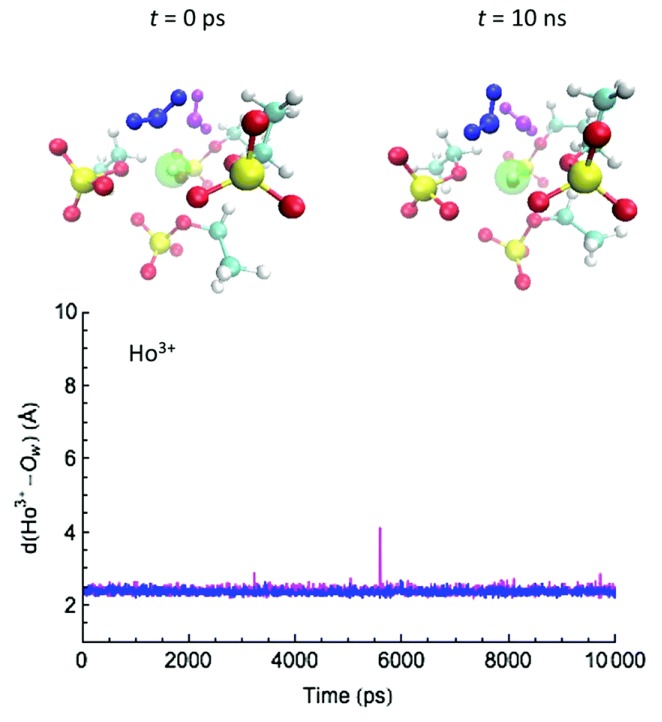
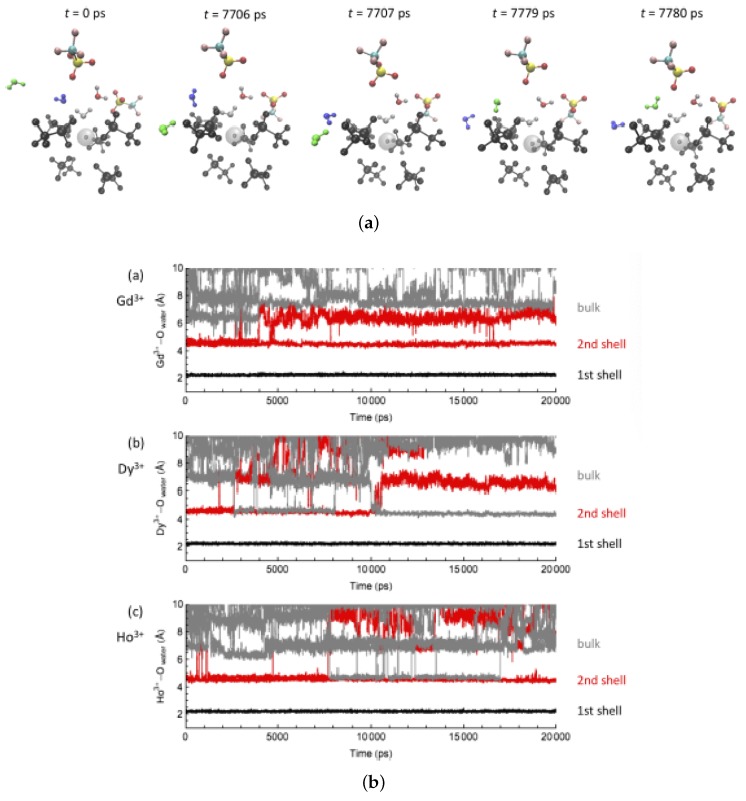
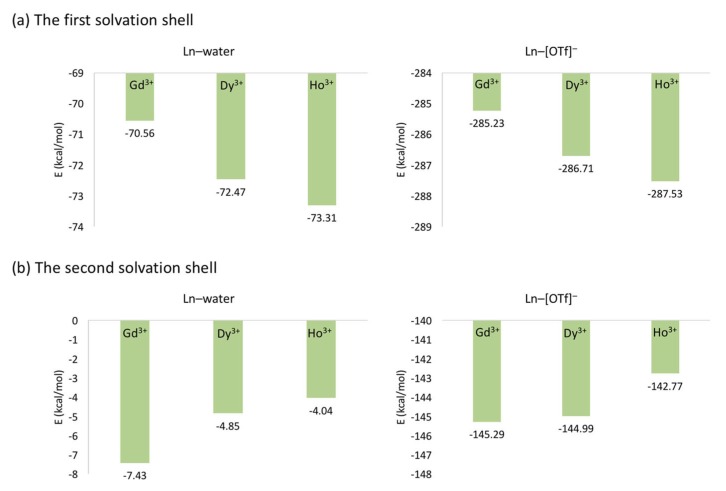
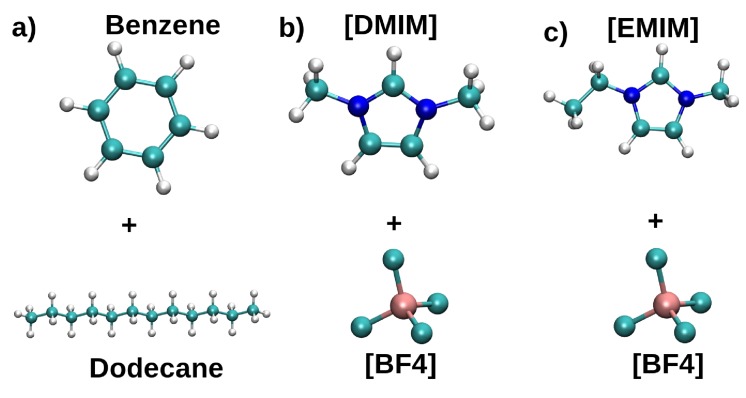
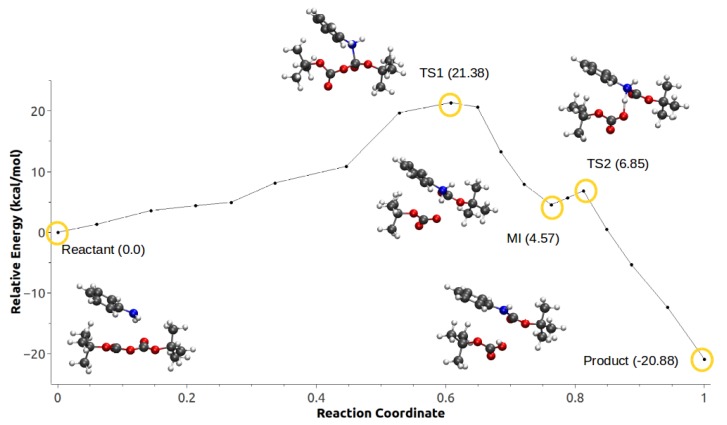
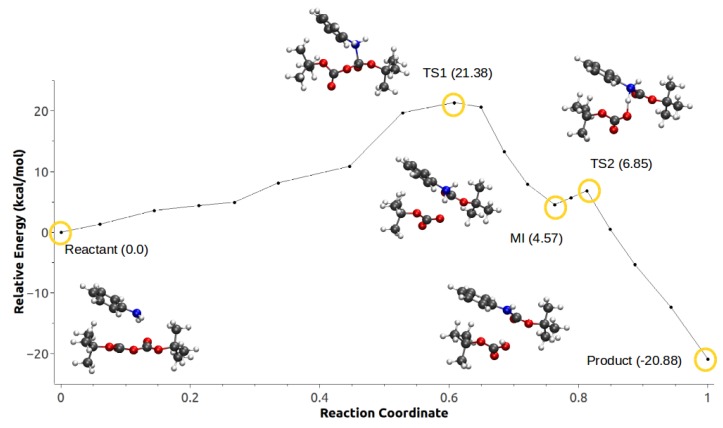
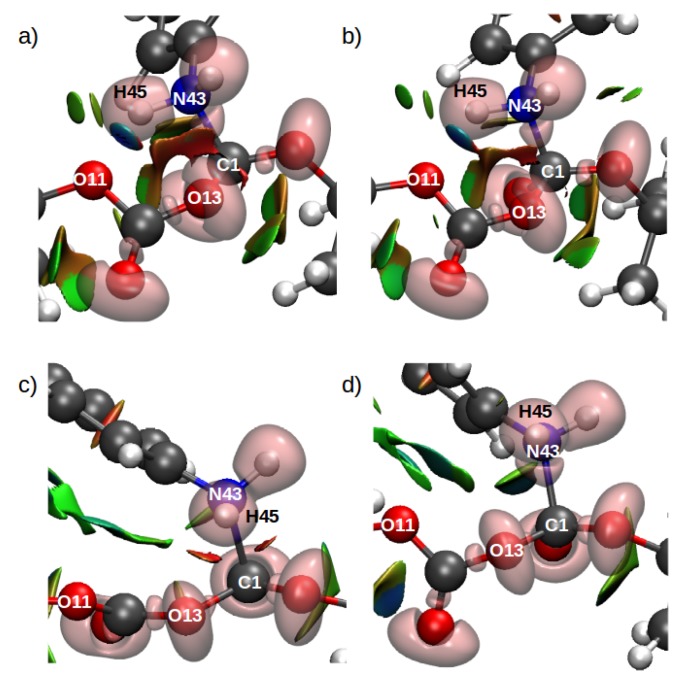
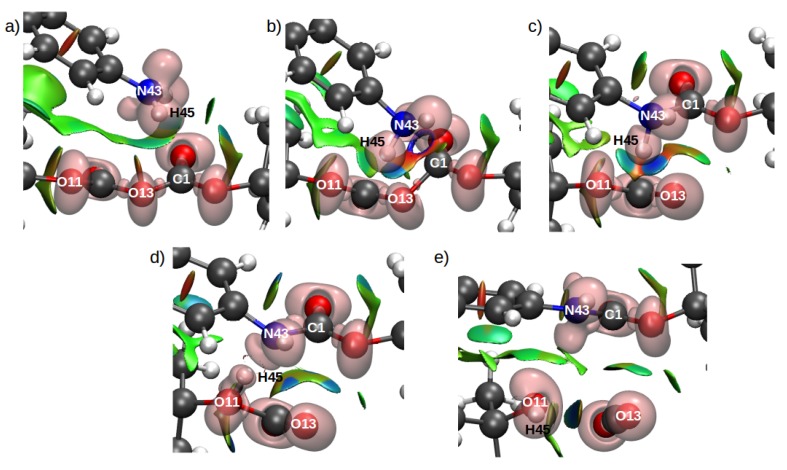
Computational simulations of ionic liquid solutions have become a useful tool to investigate various physical, chemical and catalytic properties of systems involving these solvents. Classical molecular dynamics and hybrid quantum mechanical/molecular mechanical (QM/MM) calculations of IL systems have provided significant insights at the atomic level. Here, we present a review of the development and application of the multipolar and polarizable force field AMOEBA for ionic liquid systems, termed AMOEBA-IL. The parametrization approach for AMOEBA-IL relies on the reproduction of total quantum mechanical (QM) intermolecular interaction energies and QM energy decomposition analysis. This approach has been used to develop parameters for imidazolium- and pyrrolidinium-based ILs coupled with various inorganic anions. AMOEBA-IL has been used to investigate and predict the properties of a variety of systems including neat ILs and IL mixtures, water exchange reactions on lanthanide ions in IL mixtures, IL-based liquid-liquid extraction, and effects of ILs on an aniline protection reaction.
Keywords: Computational property prediction; Ionic Liquids, Multipolar/polarizable force field, QM/MM; Molecular Dynamics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
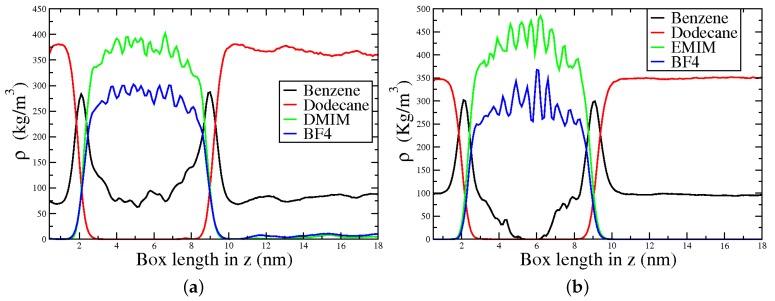
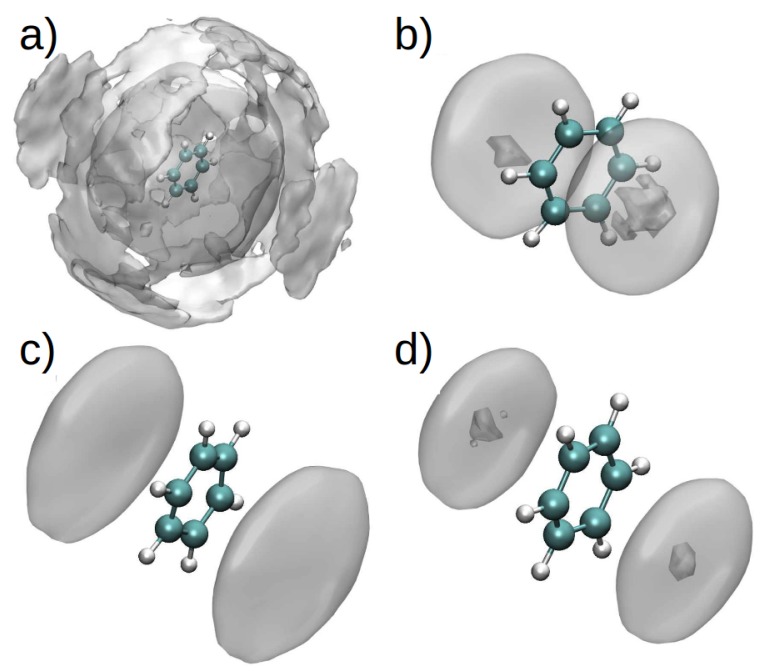
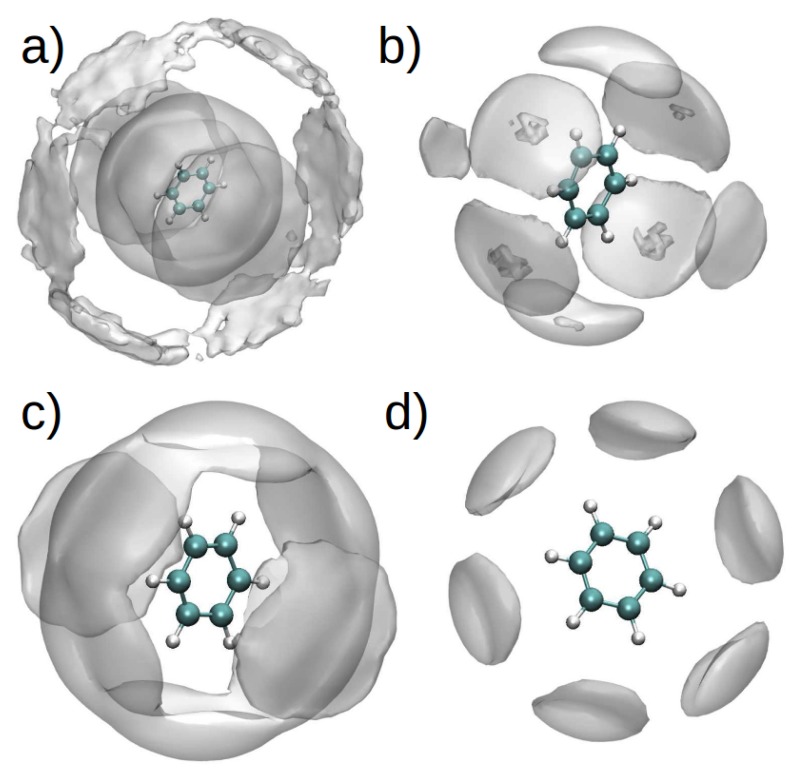
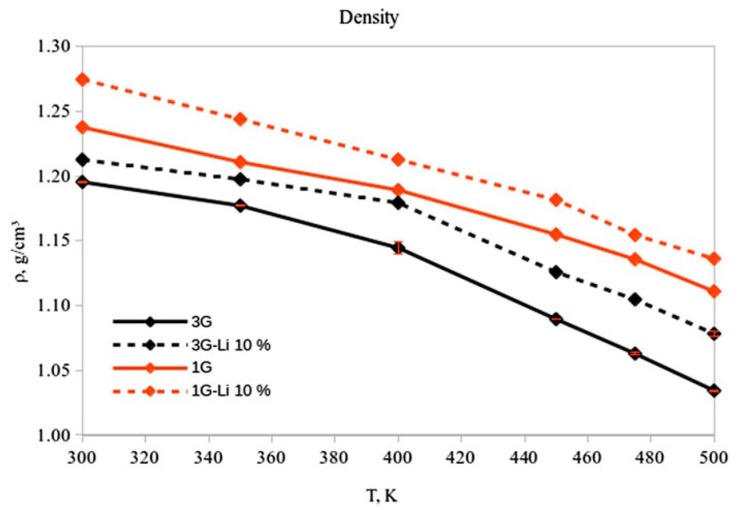
Similar articles
-
Extraction of tryptophan with ionic liquids studied with molecular dynamics simulations.J Phys Chem B. 2012 Jan 12;116(1):296-304. doi: 10.1021/jp206748z. Epub 2011 Dec 19. J Phys Chem B. 2012. PMID: 22136607
-
Mixtures of amino-acid based ionic liquids and water.J Mol Model. 2015 Sep;21(9):236. doi: 10.1007/s00894-015-2783-1. Epub 2015 Aug 16. J Mol Model. 2015. PMID: 26277480
-
Polarizable ab initio QM/MM Study of the Reaction Mechanism of N-tert-Butyloxycarbonylation of Aniline in [EMIm][BF₄].Molecules. 2018 Oct 31;23(11):2830. doi: 10.3390/molecules23112830. Molecules. 2018. PMID: 30384470 Free PMC article.
-
Chiral Ionic Liquids: Structural Diversity, Properties and Applications in Selected Separation Techniques.Int J Mol Sci. 2020 Jun 15;21(12):4253. doi: 10.3390/ijms21124253. Int J Mol Sci. 2020. PMID: 32549300 Free PMC article. Review.
-
Imidazolium-based ionic liquids for cellulose pretreatment: recent progresses and future perspectives.Appl Microbiol Biotechnol. 2017 Jan;101(2):521-532. doi: 10.1007/s00253-016-8057-8. Epub 2016 Dec 24. Appl Microbiol Biotechnol. 2017. PMID: 28012046 Review.
Cited by
-
On the Behavior of the Ethylene Glycol Components of Polydisperse Polyethylene Glycol PEG200.J Phys Chem B. 2023 Feb 9;127(5):1178-1196. doi: 10.1021/acs.jpcb.2c06773. Epub 2023 Jan 26. J Phys Chem B. 2023. PMID: 36700884 Free PMC article.
-
Perspectives in the Computational Modeling of New Generation, Biocompatible Ionic Liquids.J Phys Chem B. 2022 Jan 13;126(1):3-13. doi: 10.1021/acs.jpcb.1c09476. Epub 2022 Jan 3. J Phys Chem B. 2022. PMID: 34978449 Free PMC article. Review.
-
A Polarizable Forcefields for Glyoxal Acetals as Electrolyte Components for Lithium-Ion Batteries.ChemistryOpen. 2024 Nov;13(11):e202400134. doi: 10.1002/open.202400134. Epub 2024 Jul 31. ChemistryOpen. 2024. PMID: 39086036 Free PMC article.
-
Development of AMOEBA Polarizable Force Field for Rare-Earth La3+ Interaction with Bioinspired Ligands.J Phys Chem B. 2023 Feb 16;127(6):1367-1375. doi: 10.1021/acs.jpcb.2c07237. Epub 2023 Feb 3. J Phys Chem B. 2023. PMID: 36735638 Free PMC article.
-
The Physical Chemistry and Chemical Physics (PCCP) Section of the International Journal of Molecular Sciences in Its Publications: The First 300 Thematic Articles in the First 3 Years.Int J Mol Sci. 2021 Dec 27;23(1):241. doi: 10.3390/ijms23010241. Int J Mol Sci. 2021. PMID: 35008667 Free PMC article.
References
-
- Leach A.R. Molecular Modelling; Principles and Applications. 2nd ed. Prentice Hall; Harlow, UK: 2001.
-
- De Andrade J., Boes E.S., Stassen H. Computational Study of Room Temperature Molten Salts Composed by 1–Alkyl–3–methylimidazolium Cations—Force–Field Proposal and Validation. J. Phys. Chem. B. 2002;106:13344–13351. doi: 10.1021/jp0216629. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
