

In-silico Analysis of NF1 Missense Variants in ClinVar: Translating Variant Predictions into Variant Interpretation and Classification
- PMID: 31979111
- PMCID: PMC7037781
- DOI: 10.3390/ijms21030721
In-silico Analysis of NF1 Missense Variants in ClinVar: Translating Variant Predictions into Variant Interpretation and Classification
Abstract
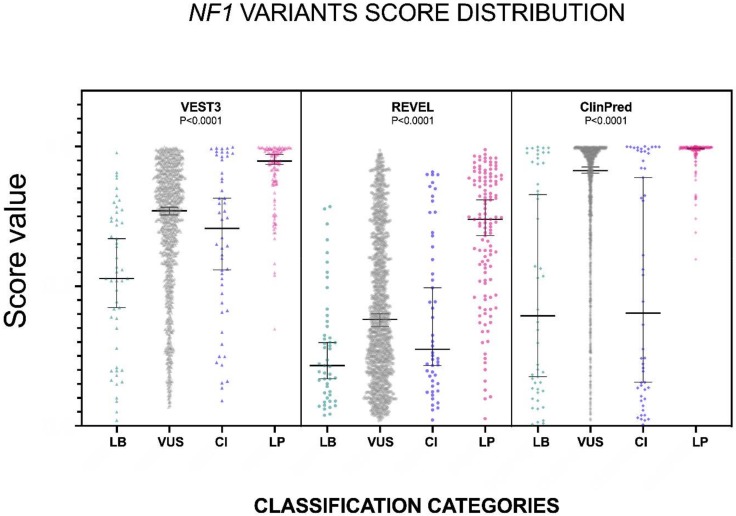
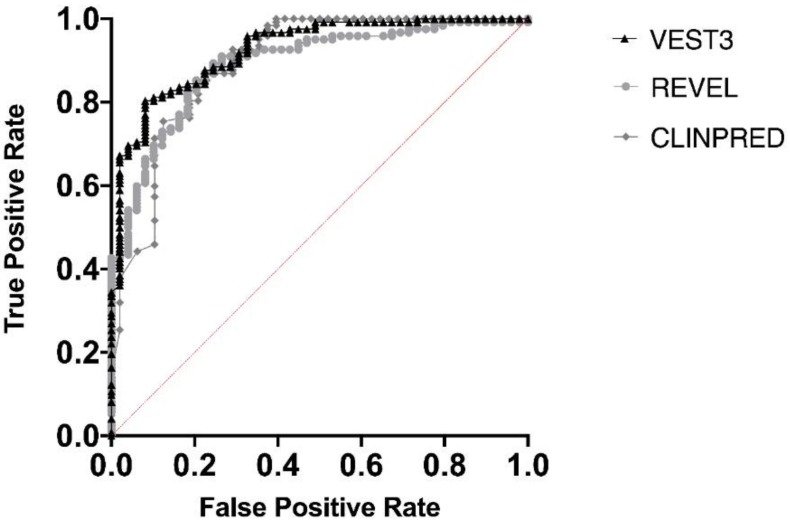
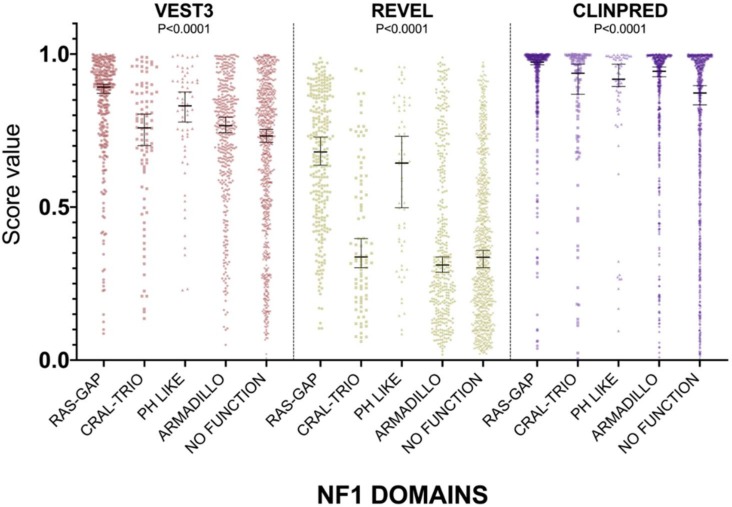
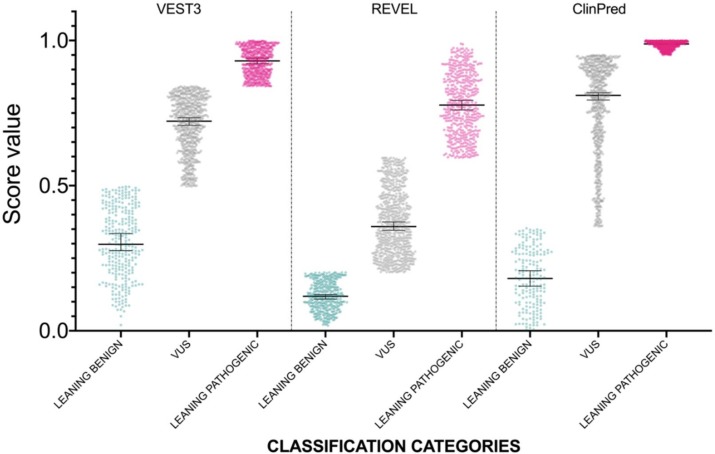
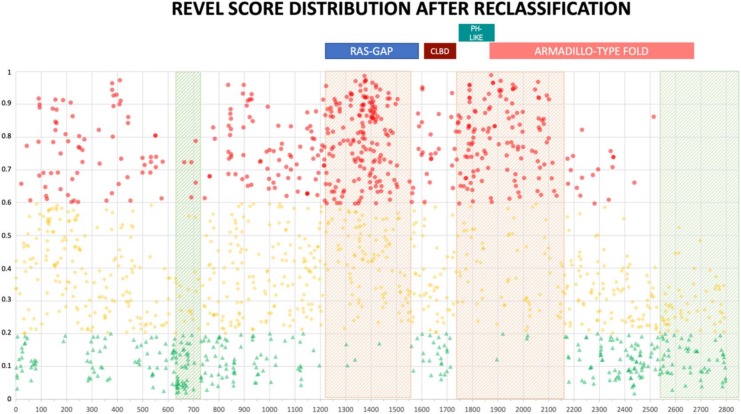
Background: With the advent of next-generation sequencing in genetic testing, predicting the pathogenicity of missense variants represents a major challenge potentially leading to misdiagnoses in the clinical setting. In neurofibromatosis type 1 (NF1), where clinical criteria for diagnosis may not be fully present until late infancy, correct assessment of variant pathogenicity is fundamental for appropriate patients' management. Methods: Here, we analyzed three different computational methods, VEST3, REVEL and ClinPred, and after extracting predictions scores for 1585 NF1 missense variants listed in ClinVar, evaluated their performances and the score distribution throughout the neurofibromin protein. Results: For all the three methods, no significant differences were present between the scores of "likely benign", "benign", and "likely pathogenic", "pathogenic" variants that were consequently collapsed into a single category. The cutoff values for pathogenicity were significantly different for the three methods and among benign and pathogenic variants for all methods. After training five different models with a subset of benign and pathogenic variants, we could reclassify variants in three sharply separated categories. Conclusions: The recently developed metapredictors, which integrate information from multiple components, after gene-specific fine-tuning, could represent useful tools for variant interpretation, particularly in genetic diseases where a clinical diagnosis can be difficult.
Keywords: ClinPred; NF1; REVEL; VEST3; missense variants; variant interpretation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Weterman M.A.J., Kuo M., Kenter S.B., Gordillo S., Karjosukarso D.W., Takase R., Bronk M., Oprescu S., van Ruissen F., Witteveen R.J.W., et al. Hypermorphic and hypomorphic AARS alleles in patients with CMT2N expand clinical and molecular heterogeneities. Hum. Mol. Genet. 2018;27:4036–4050. doi: 10.1093/hmg/ddy290. - DOI - PMC - PubMed
-
- Jamilloux Y., Lefeuvre L., Magnotti F., Martin A., Benezech S., Allatif O., Penel-Page M., Hentgen V., Sève P., Gerfaud-Valentin M., et al. Familial Mediterranean fever mutations are hypermorphic mutations that specifically decrease the activation threshold of the Pyrin inflammasome. Rheumatology (Oxford) 2018;57:100–111. doi: 10.1093/rheumatology/kex373. - DOI - PubMed
-
- Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., et al. Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;18:285–291. doi: 10.1038/nature19057. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
