

CFHR Gene Variations Provide Insights in the Pathogenesis of the Kidney Diseases Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy
- PMID: 31980588
- PMCID: PMC7003313
- DOI: 10.1681/ASN.2019050515
CFHR Gene Variations Provide Insights in the Pathogenesis of the Kidney Diseases Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy
Abstract
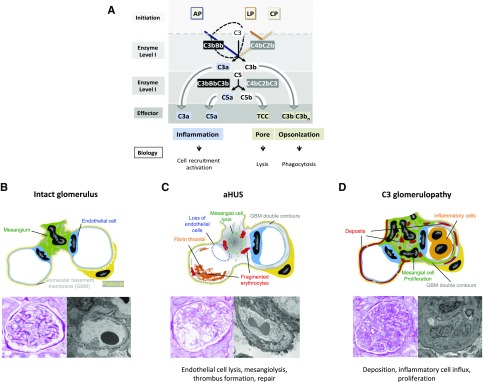
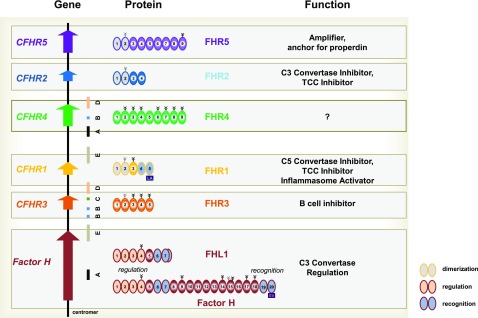
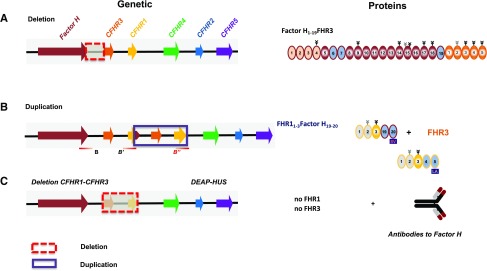
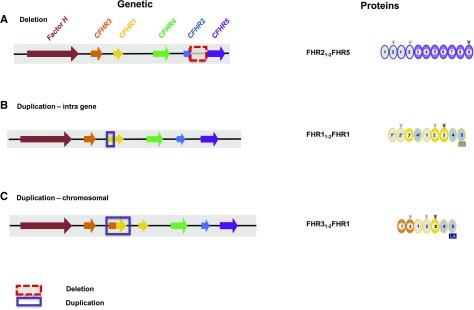
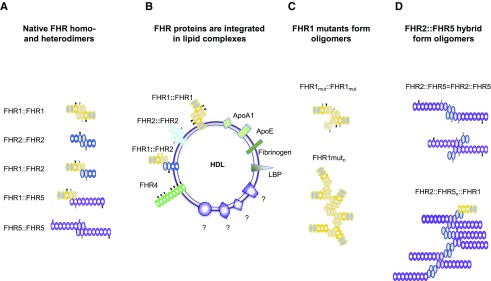
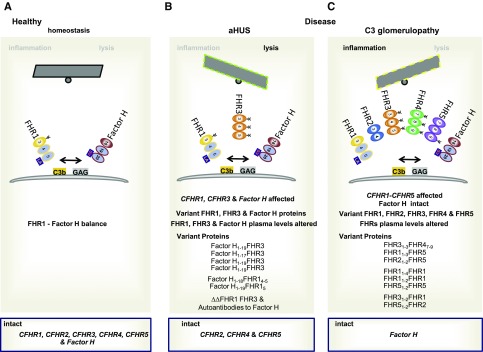
Sequence and copy number variations in the human CFHR-Factor H gene cluster comprising the complement genes CFHR1, CFHR2, CFHR3, CFHR4, CFHR5, and Factor H are linked to the human kidney diseases atypical hemolytic uremic syndrome (aHUS) and C3 glomerulopathy. Distinct genetic and chromosomal alterations, deletions, or duplications generate hybrid or mutant CFHR genes, as well as hybrid CFHR-Factor H genes, and alter the FHR and Factor H plasma repertoire. A clear association between the genetic modifications and the pathologic outcome is emerging: CFHR1, CFHR3, and Factor H gene alterations combined with intact CFHR2, CFHR4, and CFHR5 genes are reported in atypical hemolytic uremic syndrome. But alterations in each of the five CFHR genes in the context of an intact Factor H gene are described in C3 glomerulopathy. These genetic modifications influence complement function and the interplay of the five FHR proteins with each other and with Factor H. Understanding how mutant or hybrid FHR proteins, Factor H::FHR hybrid proteins, and altered Factor H, FHR plasma profiles cause pathology is of high interest for diagnosis and therapy.
Keywords: Complement Factor H related; complement; glomerular disease; glomerulonephritis; hemolytic uremic syndrome.
Copyright © 2020 by the American Society of Nephrology.
Figures
References
-
- Zipfel PF, Skerka C, Chen Q, Wiech T, Goodship T, Johnson S, et al. .: The role of complement in C3 glomerulopathy. Mol Immunol 67: 21–30, 2015. - PubMed
-
- Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, et al. .: Atypical aHUS: State of the art. Mol Immunol 67: 31–42, 2015. - PubMed
-
- Noris M, Mescia F, Remuzzi G: STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol 8: 622–633, 2012. - PubMed
-
- Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Frémeaux-Bacchi V, Kavanagh D, et al. .Conference Participants ;: Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) controversies conference. Kidney Int 91: 539–551, 2017. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
