

Cancer Cells Employ Nuclear Caspase-8 to Overcome the p53-Dependent G2/M Checkpoint through Cleavage of USP28
- PMID: 31982308
- PMCID: PMC7060810
- DOI: 10.1016/j.molcel.2019.12.023
Cancer Cells Employ Nuclear Caspase-8 to Overcome the p53-Dependent G2/M Checkpoint through Cleavage of USP28
Abstract
Cytosolic caspase-8 is a mediator of death receptor signaling. While caspase-8 expression is lost in some tumors, it is increased in others, indicating a conditional pro-survival function of caspase-8 in cancer. Here, we show that tumor cells employ DNA-damage-induced nuclear caspase-8 to override the p53-dependent G2/M cell-cycle checkpoint. Caspase-8 is upregulated and localized to the nucleus in multiple human cancers, correlating with treatment resistance and poor clinical outcome. Depletion of caspase-8 causes G2/M arrest, stabilization of p53, and induction of p53-dependent intrinsic apoptosis in tumor cells. In the nucleus, caspase-8 cleaves and inactivates the ubiquitin-specific peptidase 28 (USP28), preventing USP28 from de-ubiquitinating and stabilizing wild-type p53. This results in de facto p53 protein loss, switching cell fate from apoptosis toward mitosis. In summary, our work identifies a non-canonical role of caspase-8 exploited by cancer cells to override the p53-dependent G2/M cell-cycle checkpoint.
Keywords: G2/M checkpoint; USP28; apoptosis; cancer; caspase-8; p53.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures



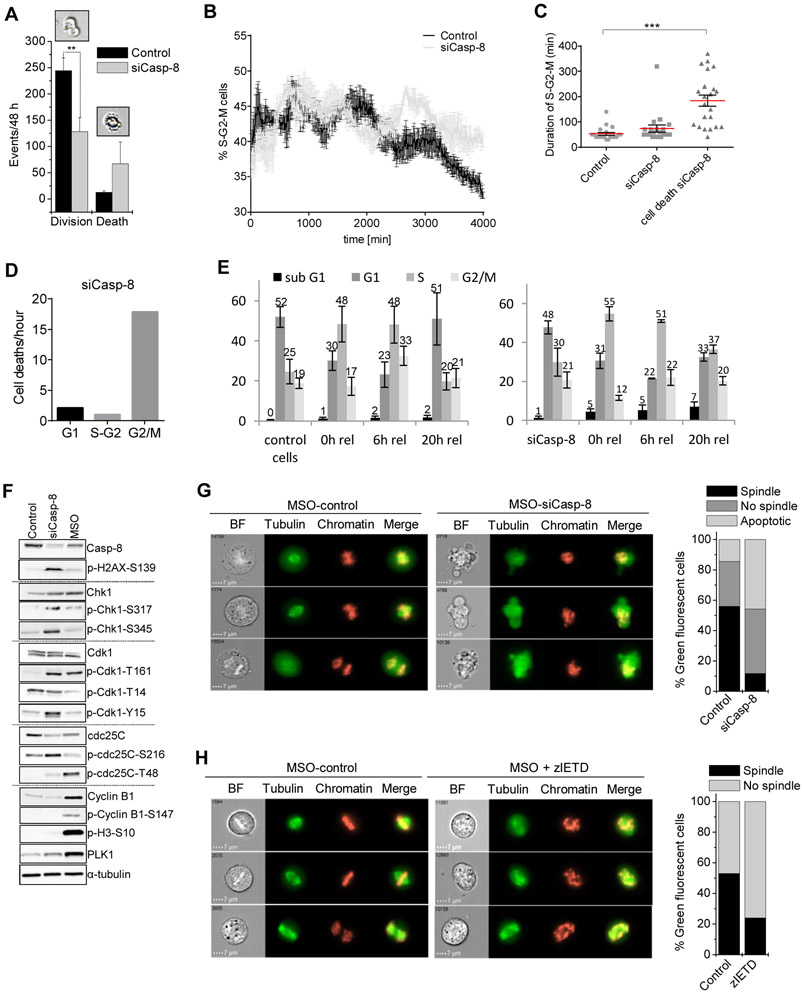


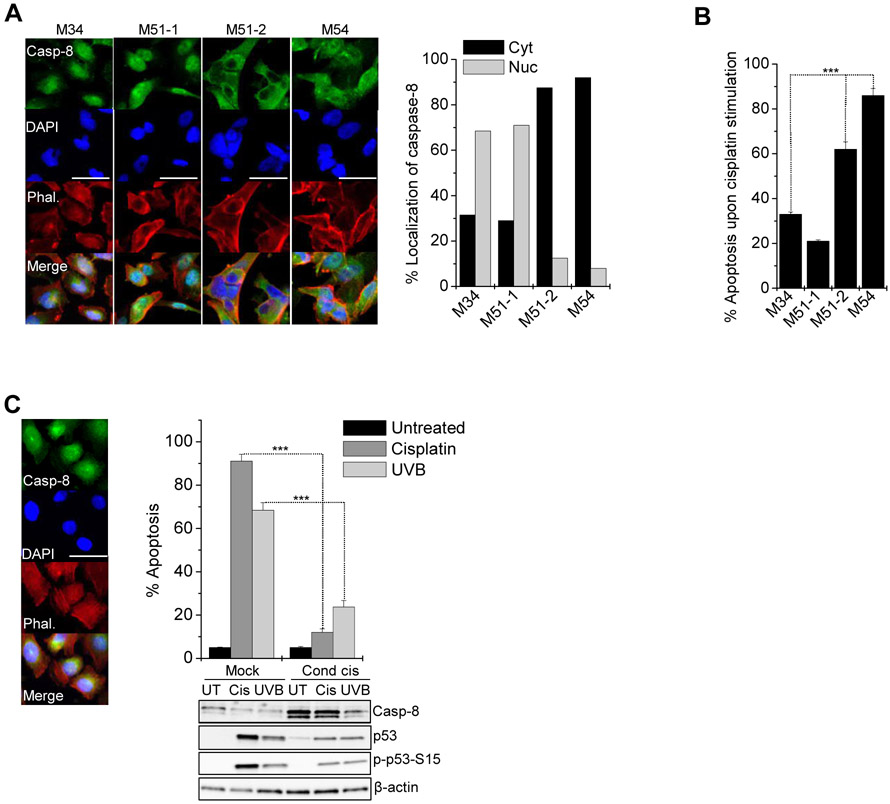
References
-
- Allende-Vega N, Dayal S, Agarwala U, Sparks A, Bourdon JC, and Saville MK (2013). p53 is activated in response to disruption of the pre-mRNA splicing machinery. Oncogene 32, 1–14. - PubMed
-
- Berglind H, Pawitan Y, Kato S, Ishioka C, and Soussi T (2008). Analysis of p53 mutation status in human cancer cell lines: a paradigm for cell line cross-contamination. Cancer Biol. Ther 7, 699–708. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
