

Identification of Lineage-Specific Transcription Factors That Prevent Activation of Hepatic Stellate Cells and Promote Fibrosis Resolution
- PMID: 31982409
- PMCID: PMC7252905
- DOI: 10.1053/j.gastro.2020.01.027
Identification of Lineage-Specific Transcription Factors That Prevent Activation of Hepatic Stellate Cells and Promote Fibrosis Resolution
Abstract
Background & aims: Development of liver fibrosis is associated with activation of quiescent hepatic stellate cells (HSCs) into collagen type I-producing myofibroblasts (activated HSCs). Cessation of liver injury often results in fibrosis resolution and inactivation of activated HSCs/myofibroblasts into a quiescent-like state (inactivated HSCs). We aimed to identify molecular features of phenotypes of HSCs from mice and humans.
Methods: We performed studies with LratCre, Ets1-floxed, Nf1-floxed, Pparγ-floxed, Gata6-floxed, Rag2-/-γc-/-, and C57/Bl6 (control) mice. Some mice were given carbon tetrachloride (CCl4) to induce liver fibrosis, with or without a peroxisome proliferator-activated receptor-γ (PPARγ) agonist. Livers from mice were analyzed by immunohistochemistry. Quiescent, activated, and inactivated HSCs were isolated from livers of Col1α1YFP mice and analyzed by chromatin immunoprecipitation and sequencing. Human HSCs were isolated from livers denied for transplantation. We compared changes in gene expression patterns and epigenetic modifications (histone H3 lysine 4 dimethylation and histone H3 lysine 27 acetylation) in primary mouse and human HSCs. Transcription factors were knocked down with small hairpin RNAs in mouse HSCs.
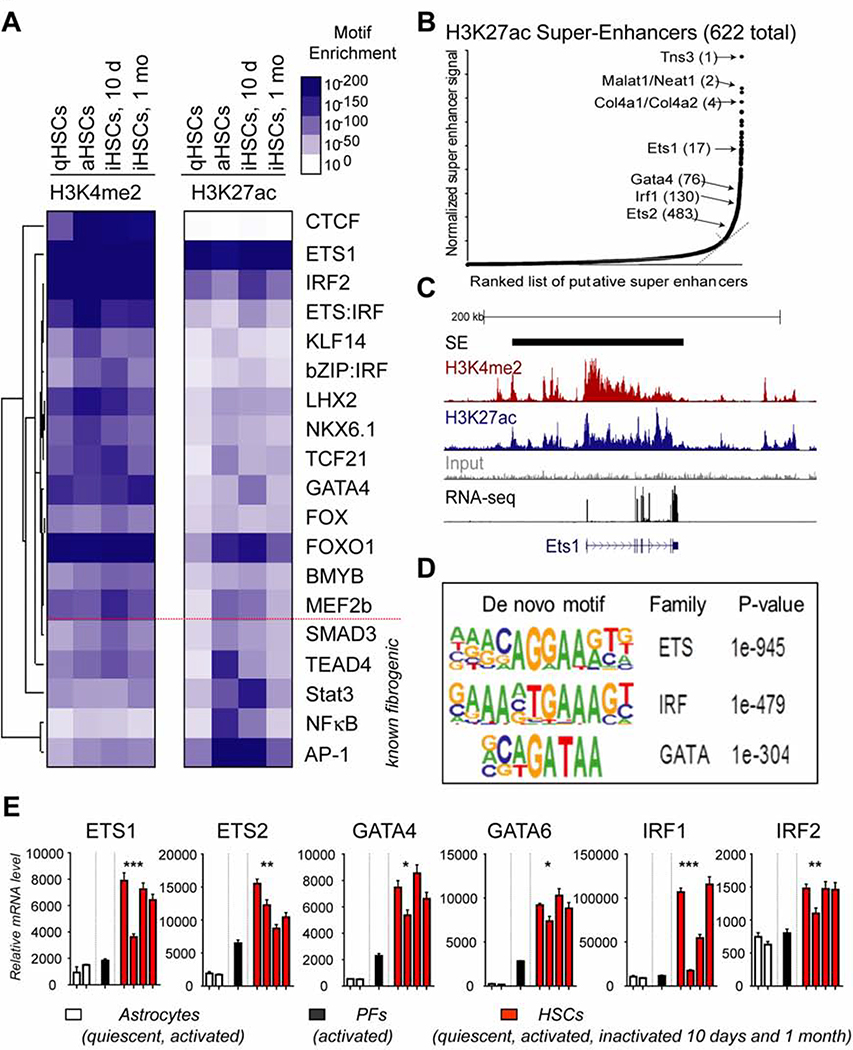
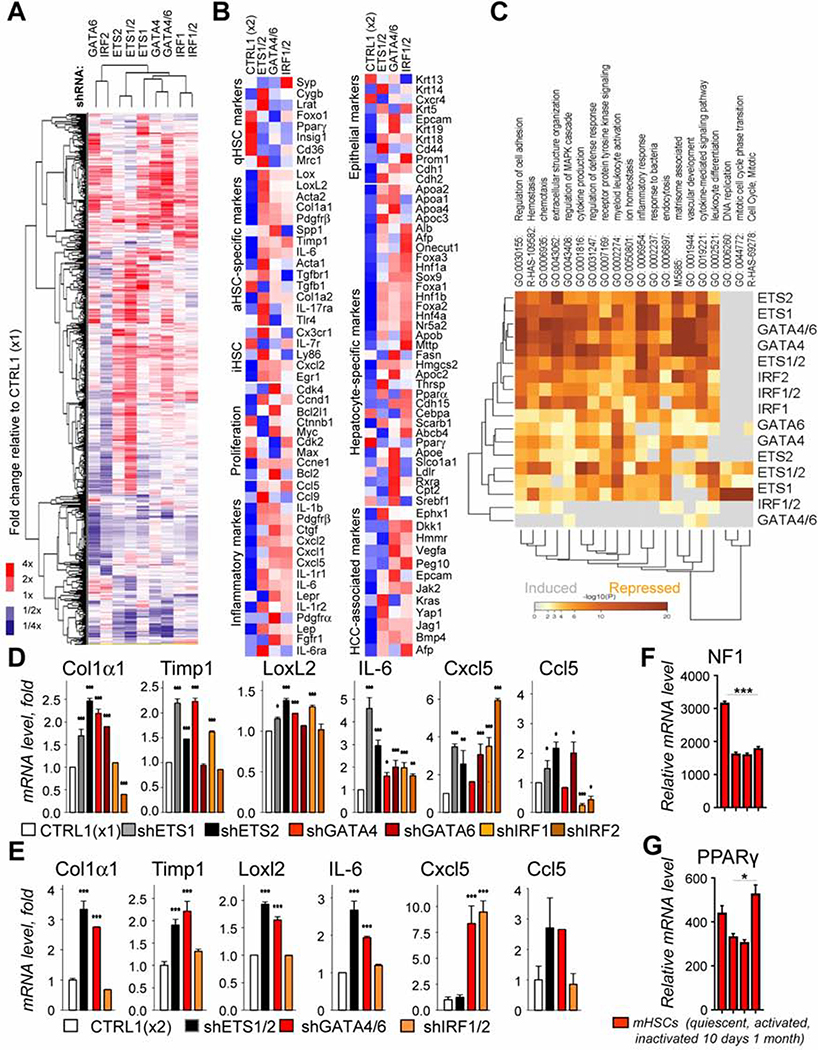
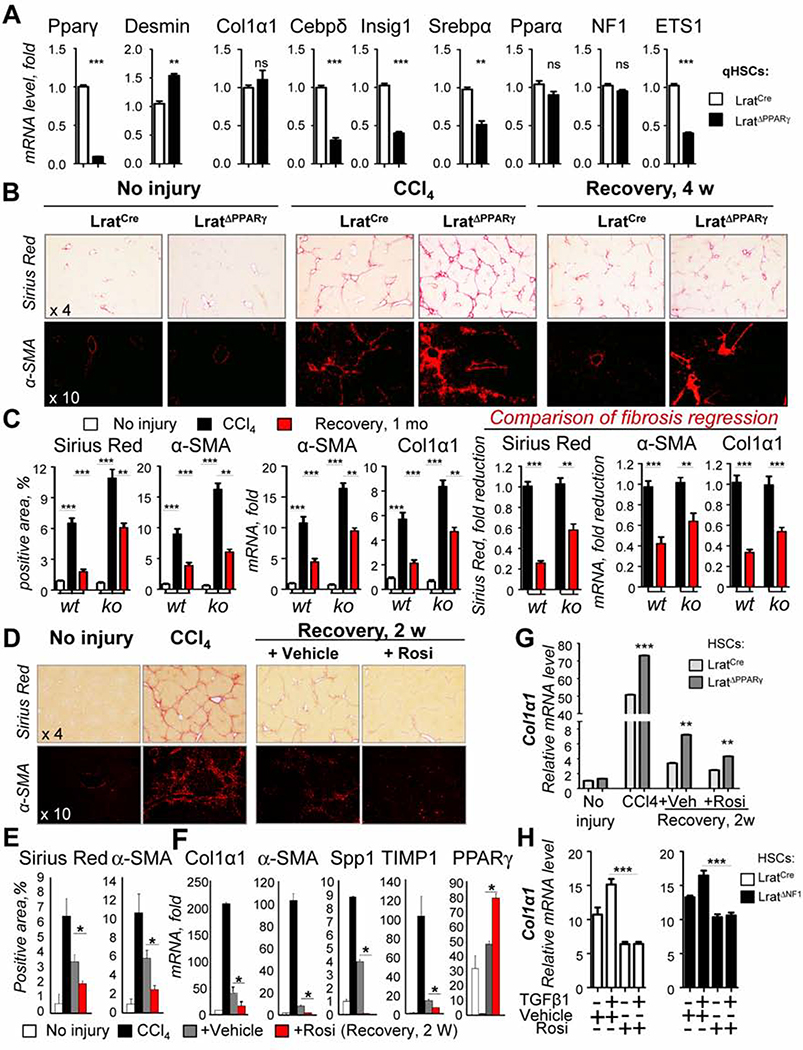
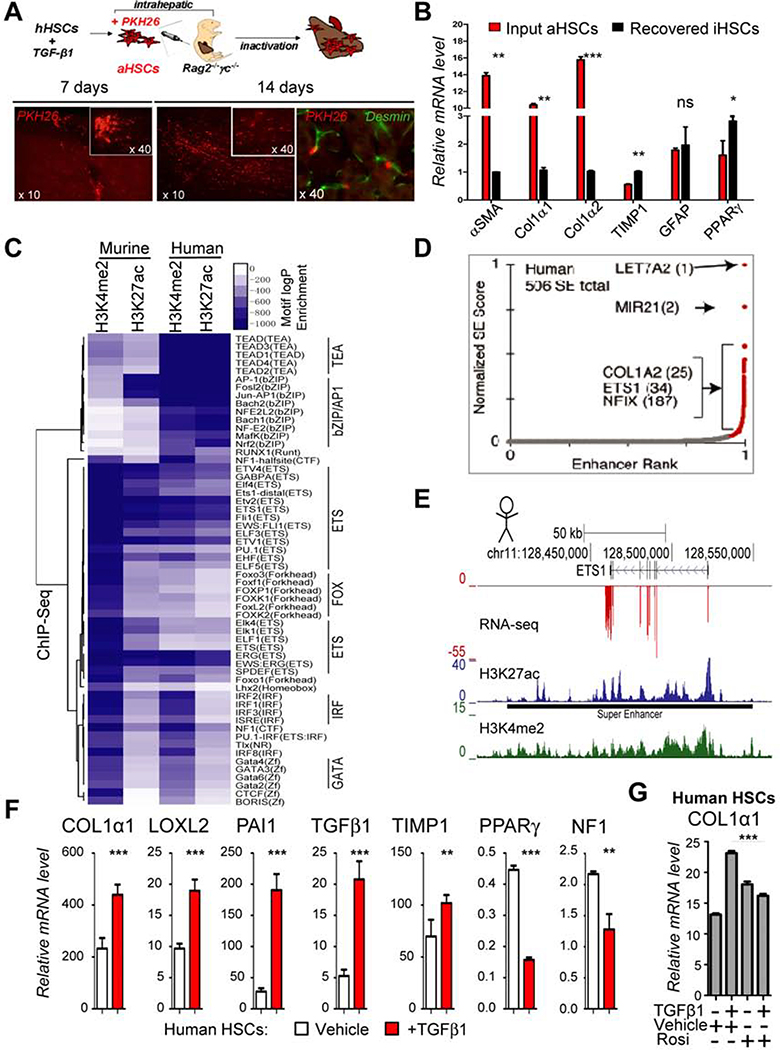
Results: Motif enrichment identified E26 transcription-specific transcription factors (ETS) 1, ETS2, GATA4, GATA6, interferon regulatory factor (IRF) 1, and IRF2 transcription factors as regulators of the mouse and human HSC lineage. Small hairpin RNA-knockdown of these transcription factors resulted in increased expression of genes that promote fibrogenesis and inflammation, and loss of HSC phenotype. Disruption of Gata6 or Ets1, or Nf1 or Pparγ (which are regulated by ETS1), increased the severity of CCl4-induced liver fibrosis in mice compared to control mice. Only mice with disruption of Gata6 or Pparγ had defects in fibrosis resolution after CCl4 administration was stopped, associated with persistent activation of HSCs. Administration of a PPARγ agonist accelerated regression of liver fibrosis after CCl4 administration in control mice but not in mice with disruption of Pparγ.
Conclusions: Phenotypes of HSCs from humans and mice are regulated by transcription factors, including ETS1, ETS2, GATA4, GATA6, IRF1, and IRF2. Activated mouse and human HSCs can revert to a quiescent-like, inactivated phenotype. We found GATA6 and PPARγ to be required for inactivation of human HSCs and regression of liver fibrosis in mice.
Keywords: Epigenetic Regulation; Inactivation of Fibrogenic Myofibroblasts; Lineage-Determining Transcription Factors; Resolution of Liver Fibrosis.
Copyright © 2020 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures



References
-
- Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007;39:311–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R37 AI043477/AI/NIAID NIH HHS/United States
- U01 AA018663/AA/NIAAA NIH HHS/United States
- I01 BX001991/BX/BLRD VA/United States
- U01 AA029019/AA/NIAAA NIH HHS/United States
- F32 DK009920/DK/NIDDK NIH HHS/United States
- R01 AA028550/AA/NIAAA NIH HHS/United States
- P30 CA023100/CA/NCI NIH HHS/United States
- I01 BX003732/BX/BLRD VA/United States
- U01 AA022614/AA/NIAAA NIH HHS/United States
- I01 BX003635/BX/BLRD VA/United States
- R01 CA199376/CA/NCI NIH HHS/United States
- R01 DK101737/DK/NIDDK NIH HHS/United States
- R56 DK088837/DK/NIDDK NIH HHS/United States
- R01 DK111866/DK/NIDDK NIH HHS/United States
- R01 DK099205/DK/NIDDK NIH HHS/United States
- P50 AA011999/AA/NIAAA NIH HHS/United States
- I01 BX002213/BX/BLRD VA/United States
- R01 AI043477/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
