

Fanconi anemia and the underlying causes of genomic instability
- PMID: 31983075
- PMCID: PMC7778457
- DOI: 10.1002/em.22358
Fanconi anemia and the underlying causes of genomic instability
Abstract
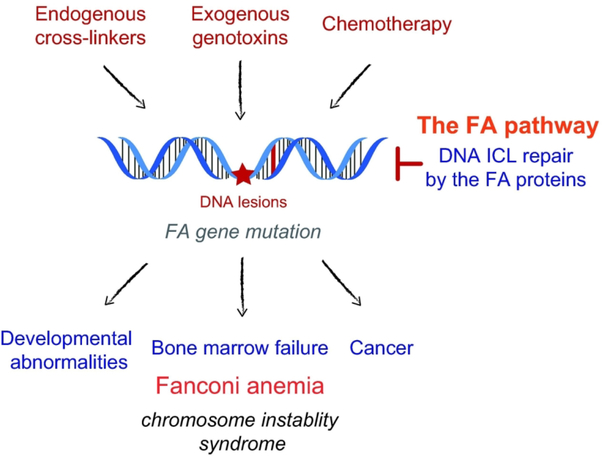
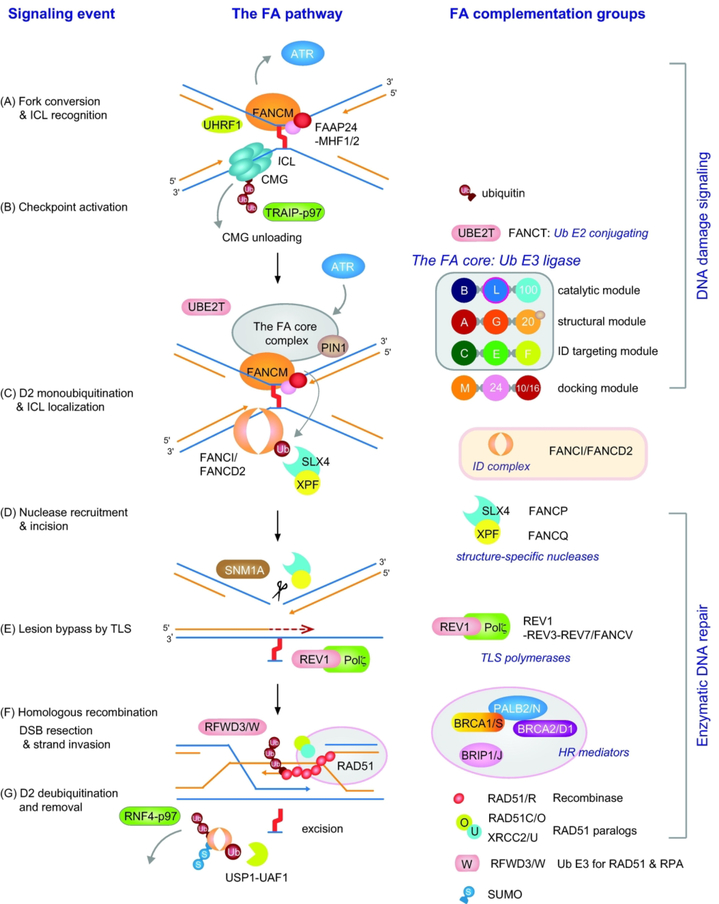
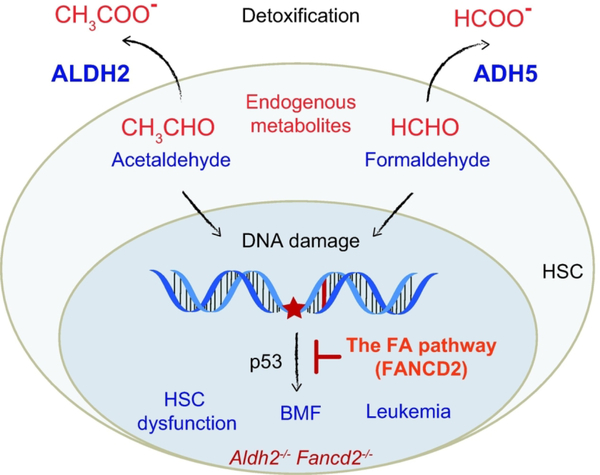
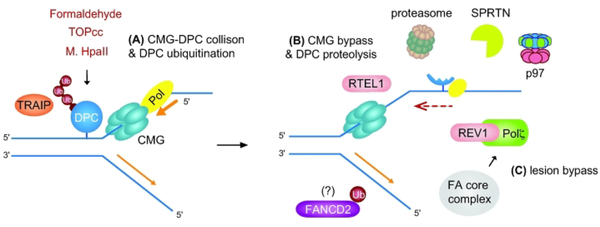
Fanconi anemia (FA) is a rare genetic disorder, characterized by birth defects, progressive bone marrow failure, and a predisposition to cancer. This devastating disease is caused by germline mutations in any one of the 22 known FA genes, where the gene products are primarily responsible for the resolution of DNA interstrand cross-links (ICLs), a type of DNA damage generally formed by cytotoxic chemotherapeutic agents. However, the identity of endogenous mutagens that generate DNA ICLs remains largely elusive. In addition, whether DNA ICLs are indeed the primary cause behind FA phenotypes is still a matter of debate. Recent genetic studies suggest that naturally occurring reactive aldehydes are a primary source of DNA damage in hematopoietic stem cells, implicating that they could play a role in genome instability and FA. Emerging lines of evidence indicate that the FA pathway constitutes a general surveillance mechanism for the genome by protecting against a variety of DNA replication stresses. Therefore, understanding the DNA repair signaling that is regulated by the FA pathway, and the types of DNA lesions underlying the FA pathophysiology is crucial for the treatment of FA and FA-associated cancers. Here, we review recent advances in our understanding of the relationship between reactive aldehydes, bone marrow dysfunction, and FA biology in the context of signaling pathways triggered during FA-mediated DNA repair and maintenance of the genomic integrity. Environ. Mol. Mutagen. 2020. © 2020 Wiley Periodicals, Inc.
Keywords: ALDH2; DNA-protein cross-link; Fanconi anemia; bone marrow failure; reactive aldehydes.
© 2020 Wiley Periodicals, Inc.
Conflict of interest statement
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
Figures
References
-
- Abel EL, Sokol RJ. 1991. A Revised Conservative Estimate of the Incidence of FAS and its Economic Impact. Alcoholism: Clinical and Experimental Research 15(3):514–524. - PubMed
-
- Alpi AF, Pace PE, Babu MM, Patel KJ. 2008. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol Cell 32(6):767–777. - PubMed
-
- Alter BP, Caruso JP, Drachtman RA, Uchida T, Velagaleti GVN, Elghetany MT. 2000. Fanconi Anemia: Myelodysplasia as a Predictor of Outcome. Cancer Genetics and Cytogenetics 117(2):125–131. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
