

Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy
- PMID: 31983221
- PMCID: PMC7004454
- DOI: 10.1161/CIRCULATIONAHA.119.037661
Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy
Abstract
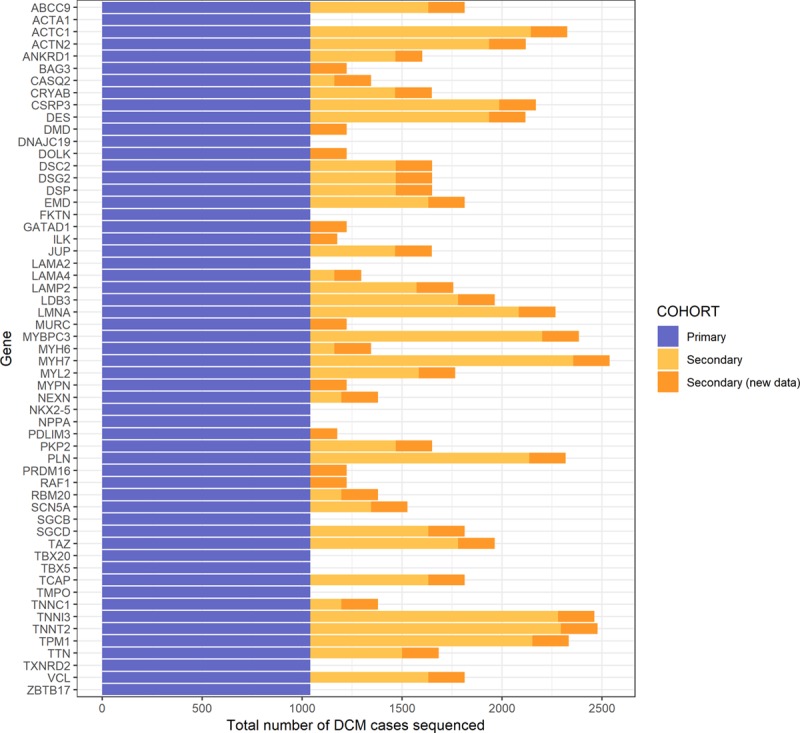
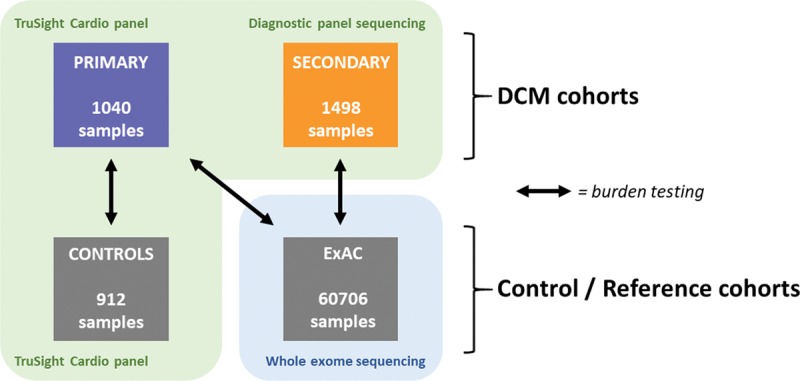
Background: Dilated cardiomyopathy (DCM) is genetically heterogeneous, with >100 purported disease genes tested in clinical laboratories. However, many genes were originally identified based on candidate-gene studies that did not adequately account for background population variation. Here we define the frequency of rare variation in 2538 patients with DCM across protein-coding regions of 56 commonly tested genes and compare this to both 912 confirmed healthy controls and a reference population of 60 706 individuals to identify clinically interpretable genes robustly associated with dominant monogenic DCM.
Methods: We used the TruSight Cardio sequencing panel to evaluate the burden of rare variants in 56 putative DCM genes in 1040 patients with DCM and 912 healthy volunteers processed with identical sequencing and bioinformatics pipelines. We further aggregated data from 1498 patients with DCM sequenced in diagnostic laboratories and the Exome Aggregation Consortium database for replication and meta-analysis.
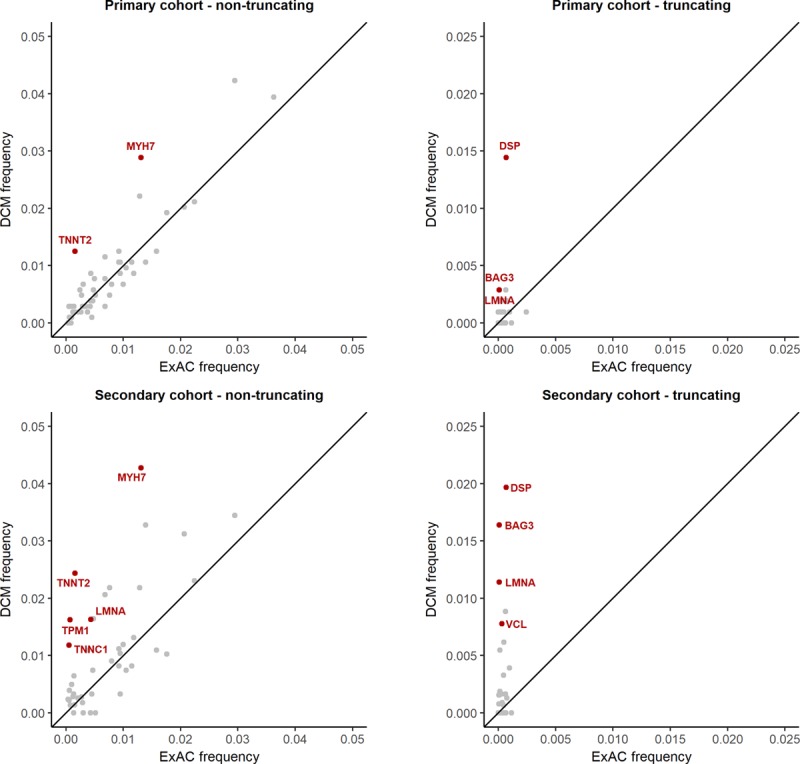
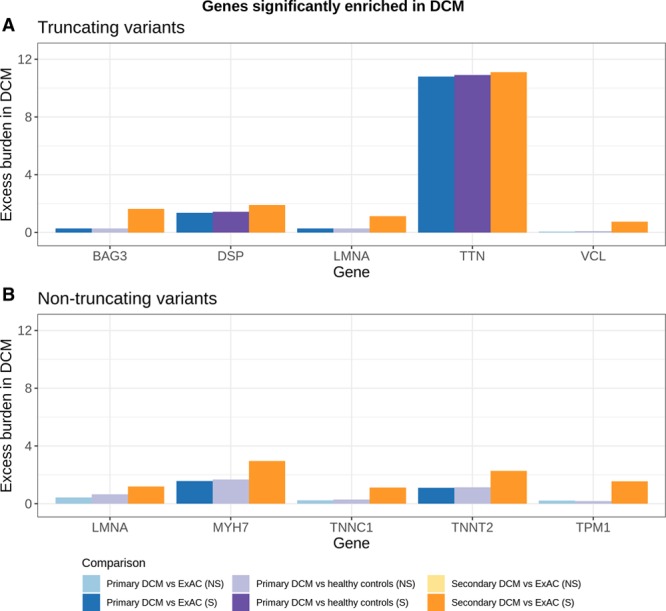
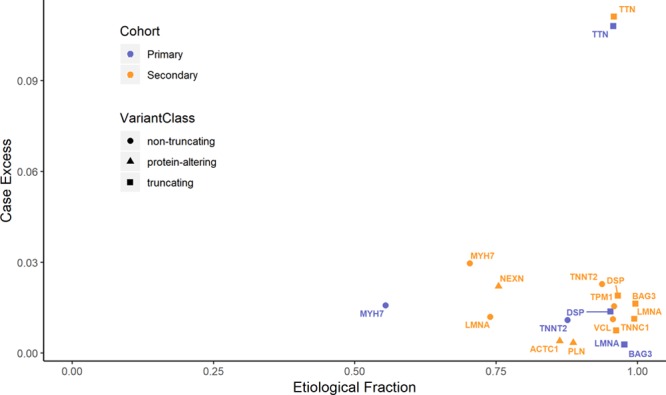
Results: Truncating variants in TTN and DSP were associated with DCM in all comparisons. Variants in MYH7, LMNA, BAG3, TNNT2, TNNC1, PLN, ACTC1, NEXN, TPM1, and VCL were significantly enriched in specific patient subsets, with the last 2 genes potentially contributing primarily to early-onset forms of DCM. Overall, rare variants in these 12 genes potentially explained 17% of cases in the outpatient clinic cohort representing a broad range of adult patients with DCM and 26% of cases in the diagnostic referral cohort enriched in familial and early-onset DCM. Although the absence of a significant excess in other genes cannot preclude a limited role in disease, such genes have limited diagnostic value because novel variants will be uninterpretable and their diagnostic yield is minimal.
Conclusions: In the largest sequenced DCM cohort yet described, we observe robust disease association with 12 genes, highlighting their importance in DCM and translating into high interpretability in diagnostic testing. The other genes analyzed here will need to be rigorously evaluated in ongoing curation efforts to determine their validity as Mendelian DCM genes but have limited value in diagnostic testing in DCM at present. This data will contribute to community gene curation efforts and will reduce erroneous and inconclusive findings in diagnostic testing.
Keywords: ExAC; Mendelian genetics; dilated cardiomyopathy; genetic testing; rare variant association testing.
Figures
References
-
- Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. doi: 10.1038/nrcardio.2013.105. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- SP/10/10/28431 /BHF_/British Heart Foundation/United Kingdom
- MC_UP_1102/19/MRC_/Medical Research Council/United Kingdom
- RG/18/9/33887/BHF_/British Heart Foundation/United Kingdom
- HICF-R6-373/DH_/Department of Health/United Kingdom
- MR/M003191/1/MRC_/Medical Research Council/United Kingdom
- RG/19/6/34387/BHF_/British Heart Foundation/United Kingdom
- RG/12/16/29939/BHF_/British Heart Foundation/United Kingdom
- 200990/A/16/Z/WT_/Wellcome Trust/United Kingdom
- NH/17/1/32725/BHF_/British Heart Foundation/United Kingdom
- MC_U120085815/MRC_/Medical Research Council/United Kingdom
- MC_UP_1102/20/MRC_/Medical Research Council/United Kingdom
- 107469/Z/15/Z/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
