

Efficacy and safety of ataluren in patients with nonsense-mutation cystic fibrosis not receiving chronic inhaled aminoglycosides: The international, randomized, double-blind, placebo-controlled Ataluren Confirmatory Trial in Cystic Fibrosis (ACT CF)
- PMID: 31983658
- PMCID: PMC9167581
- DOI: 10.1016/j.jcf.2020.01.007
Efficacy and safety of ataluren in patients with nonsense-mutation cystic fibrosis not receiving chronic inhaled aminoglycosides: The international, randomized, double-blind, placebo-controlled Ataluren Confirmatory Trial in Cystic Fibrosis (ACT CF)
Abstract
Background: Ataluren was developed for potential treatment of nonsense-mutation cystic fibrosis (CF). A previous phase 3 ataluren study failed to meet its primary efficacy endpoint, but post-hoc analyses suggested that aminoglycosides may have interfered with ataluren's action. Thus, this subsequent trial (NCT02139306) was designed to assess the efficacy and safety of ataluren in patients with nonsense-mutation CF not receiving aminoglycosides.
Methods: Eligible subjects with nonsense-mutation CF (aged ≥6 years; percent predicted (pp) FEV1 ≥40 and ≤90) from 75 sites in 16 countries were randomly assigned in double-blinded fashion to receive oral ataluren or matching placebo thrice daily for 48 weeks. The primary endpoint was absolute change in average ppFEV1 from baseline to the average of Weeks 40 and 48.
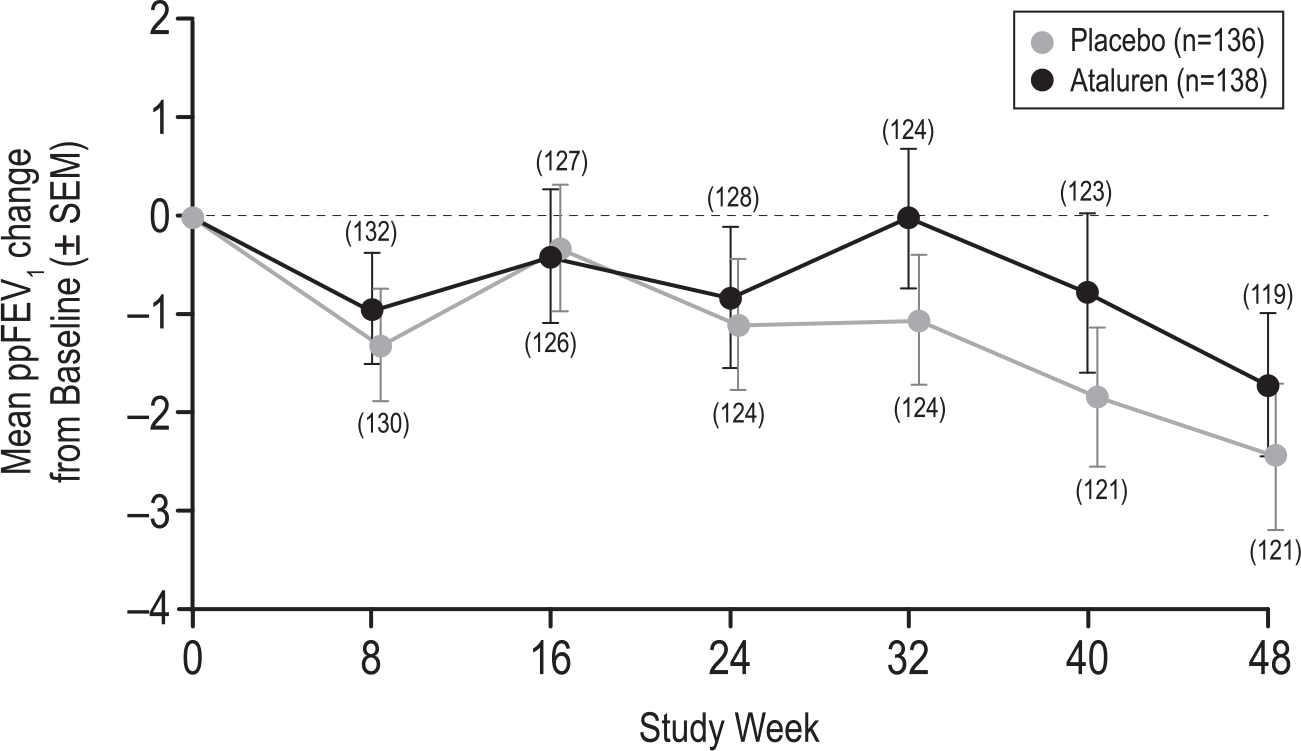
Findings: 279 subjects were enrolled; 138 subjects in the ataluren arm and 136 in the placebo arm were evaluable for efficacy. Absolute ppFEV1 change from baseline did not differ significantly between the ataluren and placebo groups at Week 40 (-0.8 vs -1.8) or Week 48 (-1.7 vs -2.4). Average ppFEV1 treatment difference from baseline to Weeks 40 and 48 was 0.6 (95% CI -1.3, 2.5; p = 0.54). Pulmonary exacerbation rate per 48 weeks was not significantly different (ataluren 0.95 vs placebo 1.13; rate ratio p = 0.40). Safety was similar between groups. No life-threatening adverse events or deaths were reported.
Interpretation: Neither ppFEV1 change nor pulmonary exacerbation rate over 48 weeks were statistically different between ataluren and placebo groups. Development of a nonsense-mutation CF therapy remains elusive.
Keywords: Ataluren; Clinical trial; Cystic fibrosis.
Copyright © 2020. Published by Elsevier B.V.
Conflict of interest statement
Declaration of Competing Interest JM is an employee of PTC Therapeutics, the funder of this clinical trial, and holds financial interests in the company. MWK, SMR, and DRV received compensation for consultant services from PTC Therapeutics prior to and/or during this study. EK, MW, IS-G, and KDB received compensation for travel expenses for meetings related to the study. All other authors declare no competing interests.
Figures

References
-
- Cystic Fibrosis Foundation Patient Registry. 2017 annual data report to the center directors. Bethesda (MD): Cystic Fibrosis Foundation; 2018.
-
- Shoshani T, Augarten A, Gazit E, Bashan N, Yahav Y, Rivlin Y, Tal A, Seret H, Yaar L, Kerem E, et al. Association of a nonsense mutation (W1282X), the most common mutation in the Ashkenazi Jewish cystic fibrosis patients in Israel, with presentation of severe disease. Am J Hum Genet 1992;50(1):222–8. - PMC - PubMed
-
- Cystic Fibrosis Genotype-Phenotype Consortium Correlation between genotype and phenotype in patients with cystic fibrosis. N Engl J Med 1993;329(18):1308–13. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
