

The SAMPL6 SAMPLing challenge: assessing the reliability and efficiency of binding free energy calculations
- PMID: 31984465
- PMCID: PMC7282318
- DOI: 10.1007/s10822-020-00290-5
The SAMPL6 SAMPLing challenge: assessing the reliability and efficiency of binding free energy calculations
Abstract
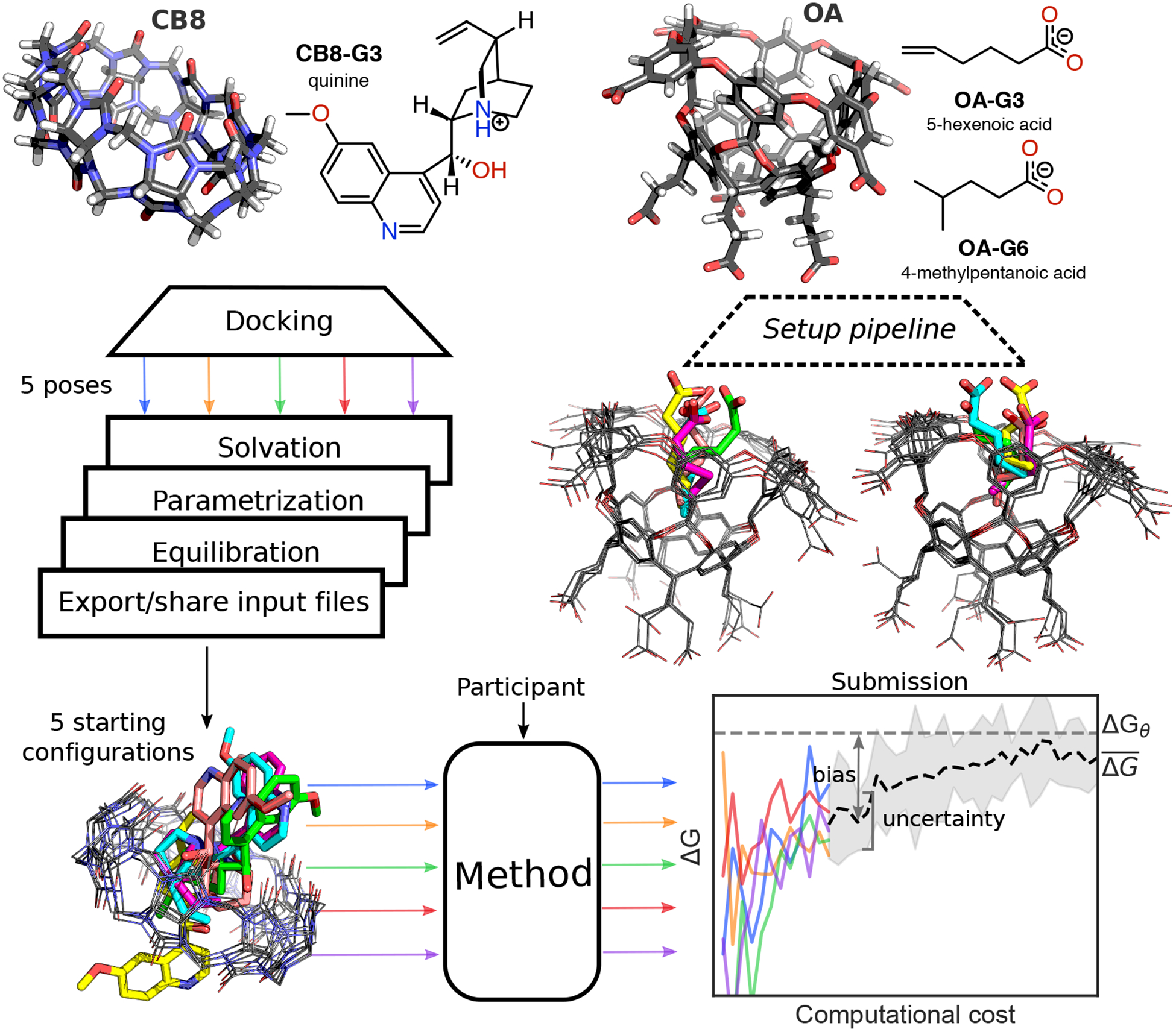
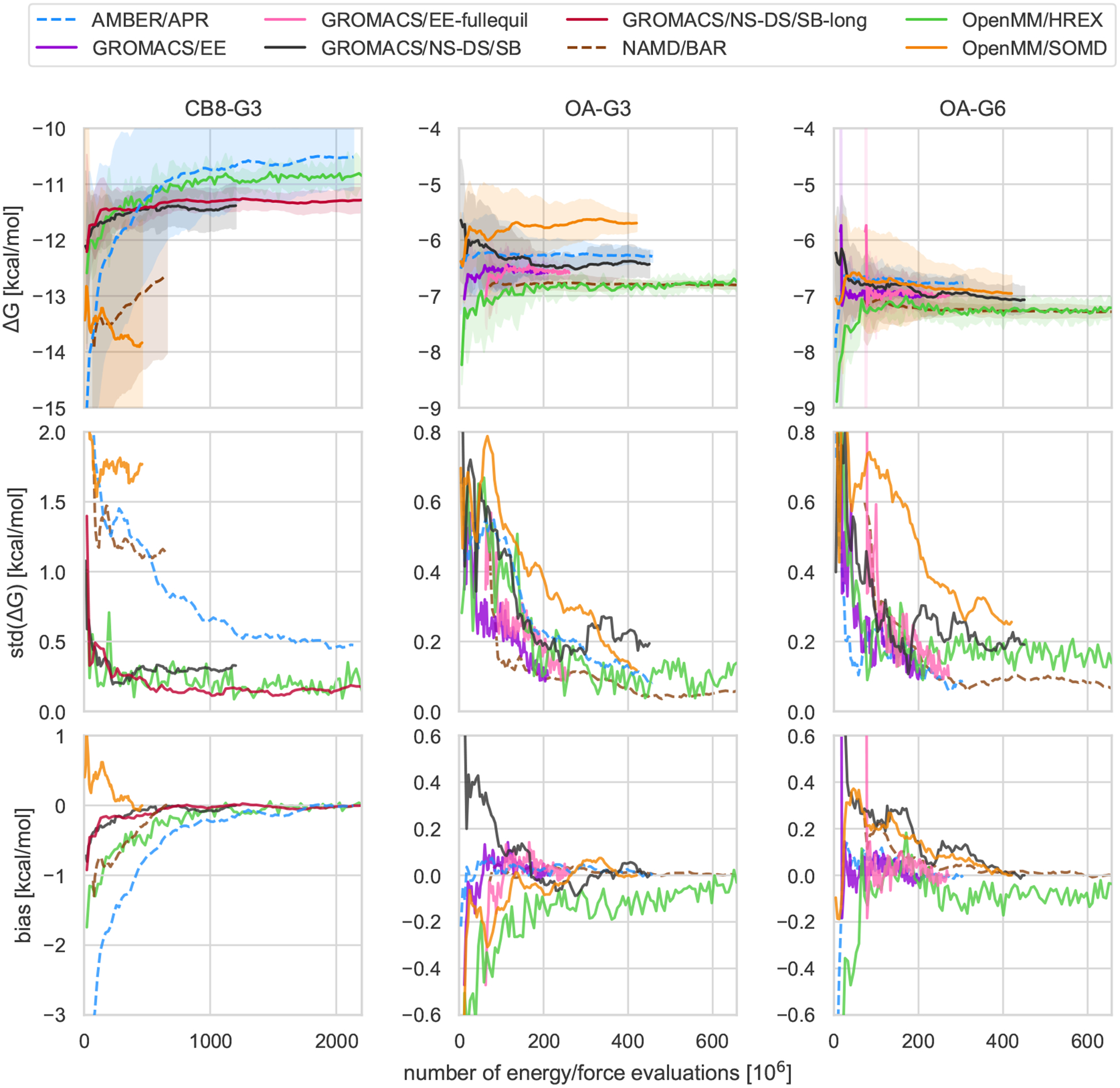
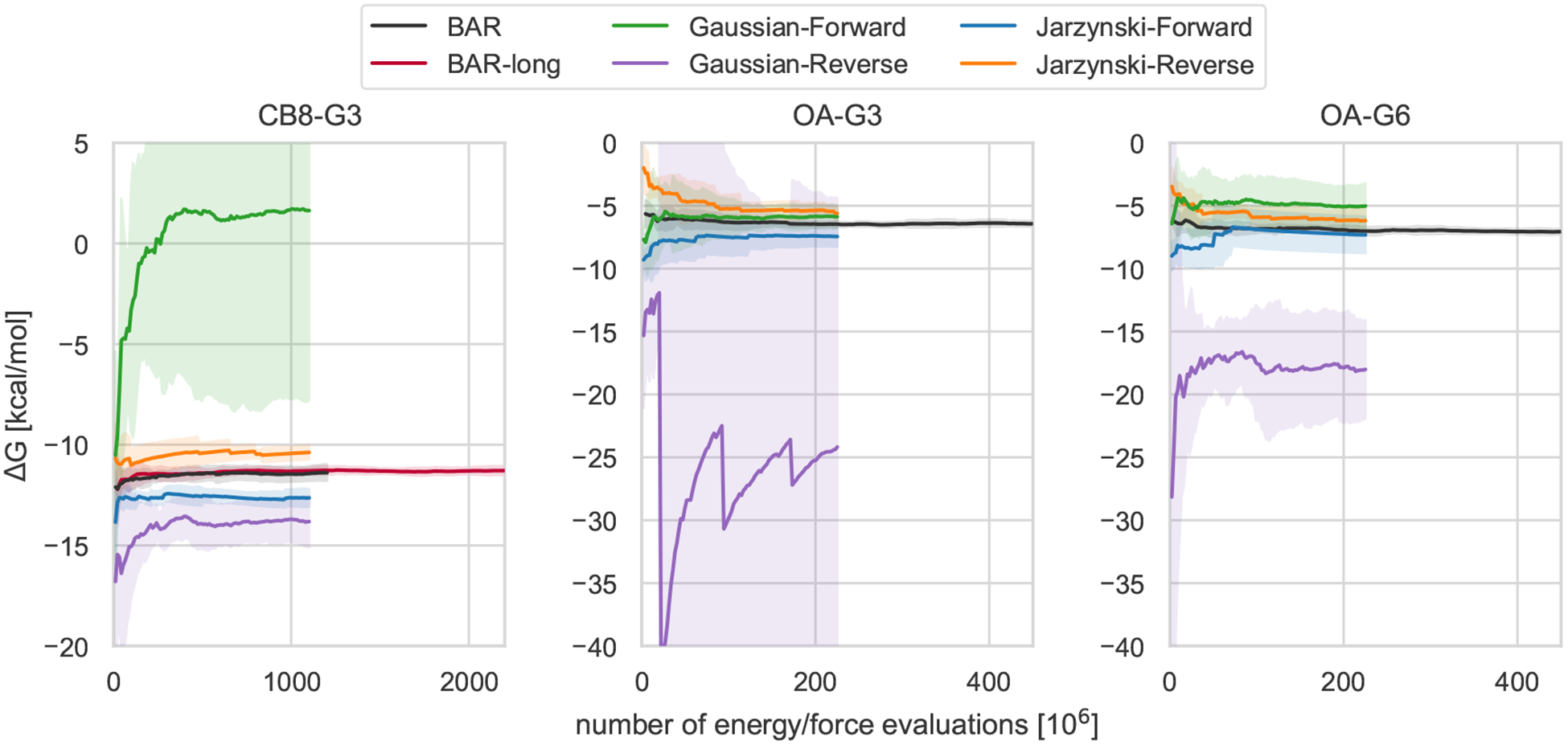
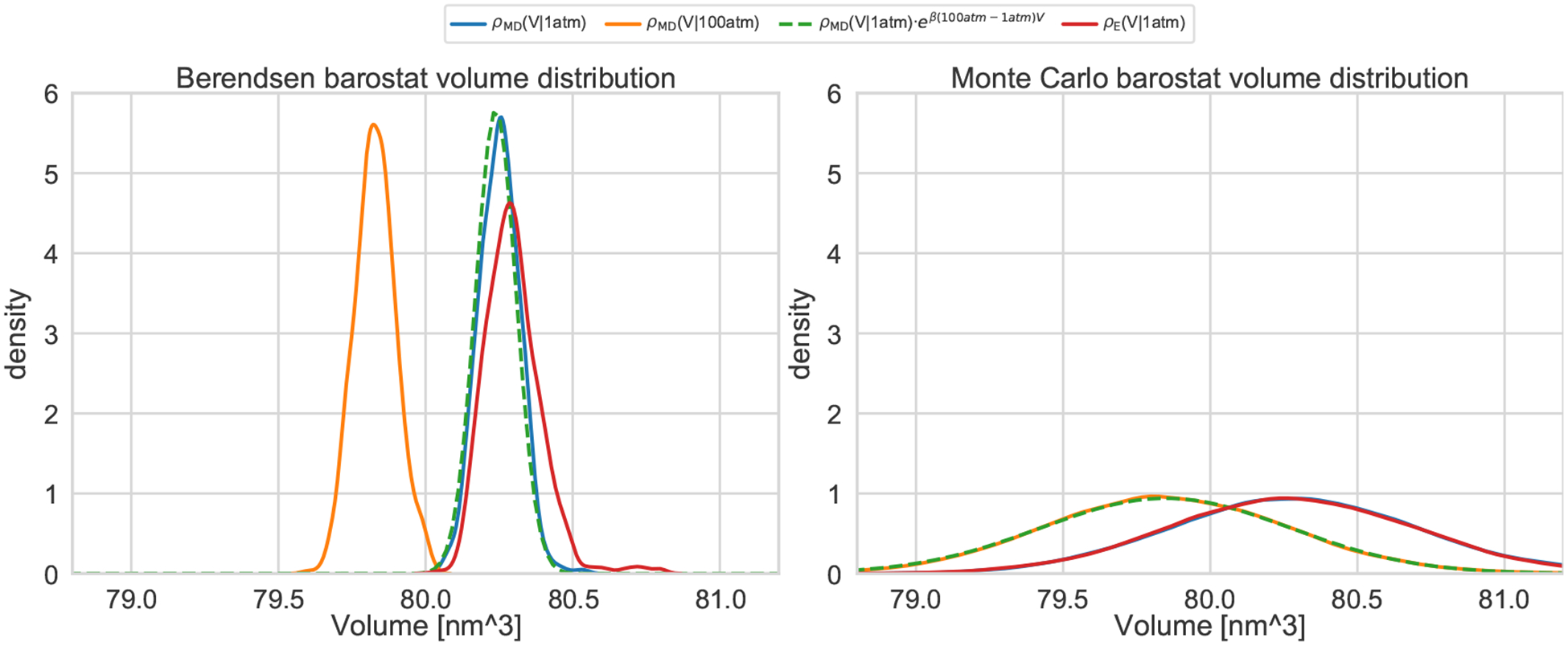
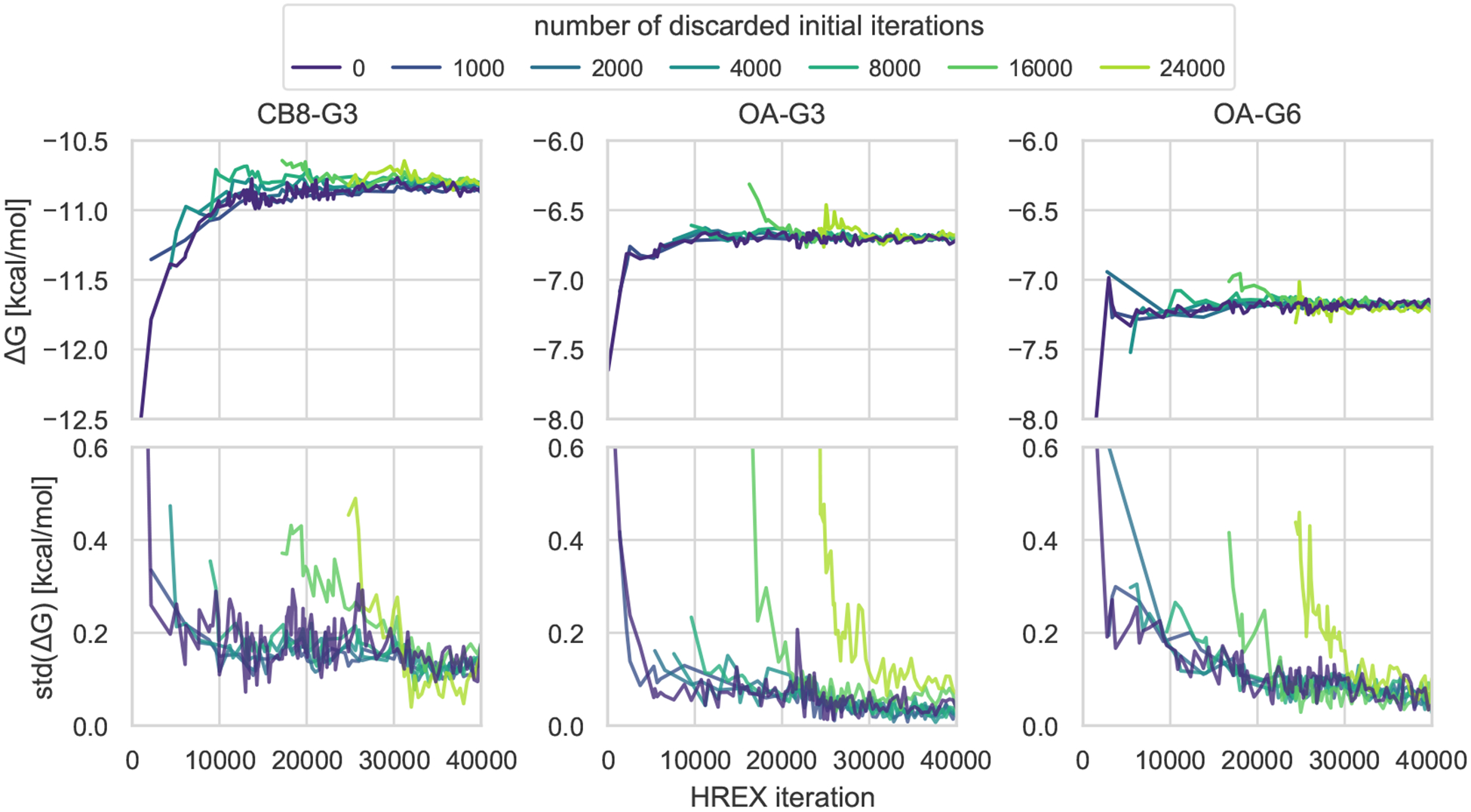
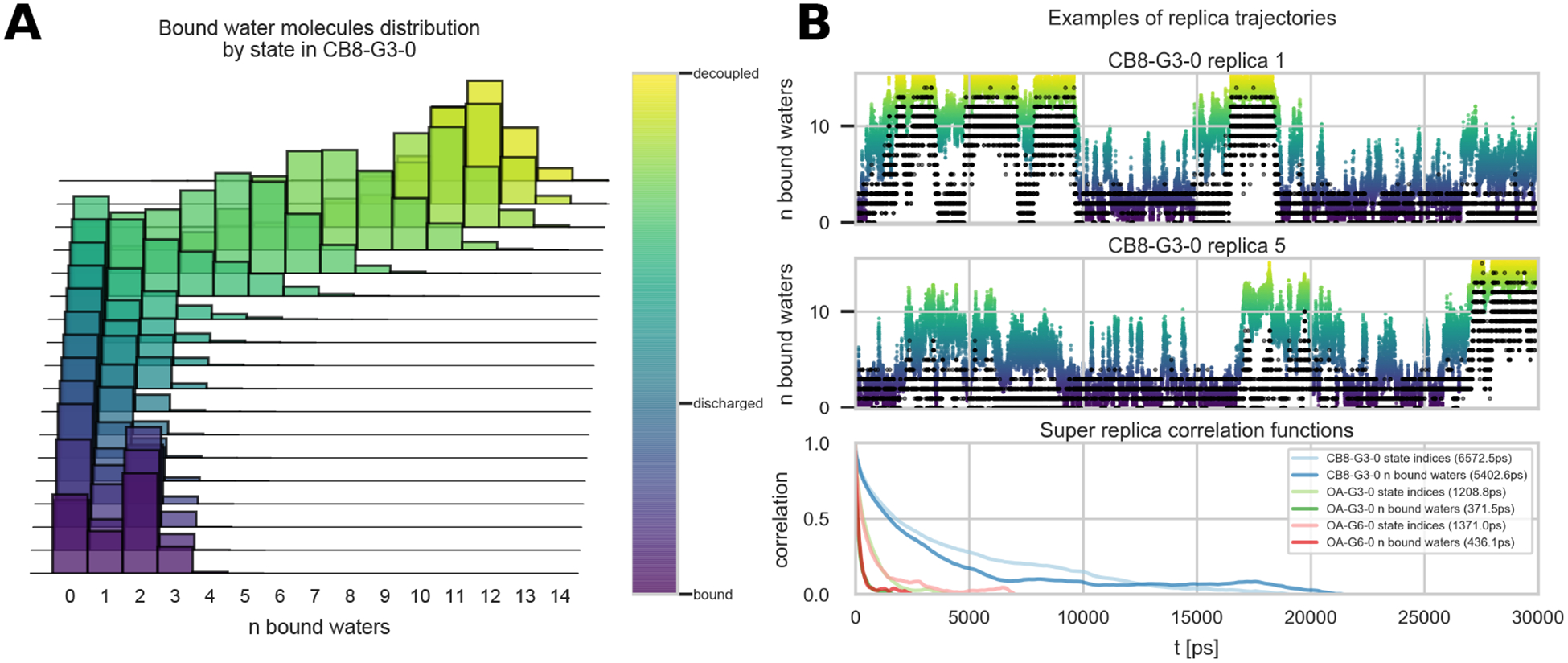
Approaches for computing small molecule binding free energies based on molecular simulations are now regularly being employed by academic and industry practitioners to study receptor-ligand systems and prioritize the synthesis of small molecules for ligand design. Given the variety of methods and implementations available, it is natural to ask how the convergence rates and final predictions of these methods compare. In this study, we describe the concept and results for the SAMPL6 SAMPLing challenge, the first challenge from the SAMPL series focusing on the assessment of convergence properties and reproducibility of binding free energy methodologies. We provided parameter files, partial charges, and multiple initial geometries for two octa-acid (OA) and one cucurbit[8]uril (CB8) host-guest systems. Participants submitted binding free energy predictions as a function of the number of force and energy evaluations for seven different alchemical and physical-pathway (i.e., potential of mean force and weighted ensemble of trajectories) methodologies implemented with the GROMACS, AMBER, NAMD, or OpenMM simulation engines. To rank the methods, we developed an efficiency statistic based on bias and variance of the free energy estimates. For the two small OA binders, the free energy estimates computed with alchemical and potential of mean force approaches show relatively similar variance and bias as a function of the number of energy/force evaluations, with the attach-pull-release (APR), GROMACS expanded ensemble, and NAMD double decoupling submissions obtaining the greatest efficiency. The differences between the methods increase when analyzing the CB8-quinine system, where both the guest size and correlation times for system dynamics are greater. For this system, nonequilibrium switching (GROMACS/NS-DS/SB) obtained the overall highest efficiency. Surprisingly, the results suggest that specifying force field parameters and partial charges is insufficient to generally ensure reproducibility, and we observe differences between seemingly converged predictions ranging approximately from 0.3 to 1.0 kcal/mol, even with almost identical simulations parameters and system setup (e.g., Lennard-Jones cutoff, ionic composition). Further work will be required to completely identify the exact source of these discrepancies. Among the conclusions emerging from the data, we found that Hamiltonian replica exchange-while displaying very small variance-can be affected by a slowly-decaying bias that depends on the initial population of the replicas, that bidirectional estimators are significantly more efficient than unidirectional estimators for nonequilibrium free energy calculations for systems considered, and that the Berendsen barostat introduces non-negligible artifacts in expanded ensemble simulations.
Keywords: Binding affinity; Cucurbit[8]uril; Free energy calculations; Host–guest; Octa-acid; SAMPL6; Sampling.
Conflict of interest statement
Disclosures
JDC was a member of the Scientific Advisory Board for Schrödinger, LLC during part of this study. JDC and DLM are current members of the Scientific Advisory Board of OpenEye Scientific Software. The Chodera laboratory receives or has received funding from multiple sources, including the National Institutes of Health, the National Science Foundation, the Parker Institute for Cancer Immunotherapy, Relay Therapeutics, Entasis Therapeutics, Silicon Therapeutics, EMD Serono (Merck KGaA), AstraZeneca, Vir Biotechnology, XtalPi, the Molecular Sciences Software Institute, the Starr Cancer Consortium, the Open Force Field Consortium, Cycle for Survival, a Louis V. Gerstner Young Investigator Award, and the Sloan Kettering Institute. A complete funding history for the Chodera lab can be found at
Figures
References
-
- Shirts MR, Mobley DL, Brown SP. Free-energy calculations in structure-based drug design. Drug design: structure- and ligand-based approaches. 2010; p. 61–86.
-
- Kuhn B, Tichý M, Wang L, Robinson S, Martin RE, Kuglstatter A, Benz J. Prospective evaluation of free energy calculations for the prioritization of cathepsin L inhibitors. Journal of medicinal chemistry. 2017; 60(6):2485–2497. - PubMed
-
- Ciordia M, Pérez-Benito L, Delgado F, Trabanco AA, Tresadern G. Application of free energy perturbation for the design of BACE1 inhibitors. Journal of Chemical information and modeling. 2016; 56(9):1856–1871. - PubMed
-
- Schindler C, Rippmann F, Kuhn D. Relative binding affinity prediction of farnesoid X receptor in the D3R Grand Challenge 2 using FEP+. Journal of computer-aided molecular design. 2018; 32(1):265–272. - PubMed
-
- Wang L, Wu Y, Deng Y, Kim B, Pierce L, Krilov G, Lupyan D, Robinson S, Dahlgren MK, Greenwood J, et al. Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. Journal of the American Chemical Society. 2015; 137(7):2695–2703. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
