

Immunometabolism in the development of rheumatoid arthritis
- PMID: 31984519
- PMCID: PMC7047523
- DOI: 10.1111/imr.12838
Immunometabolism in the development of rheumatoid arthritis
Abstract
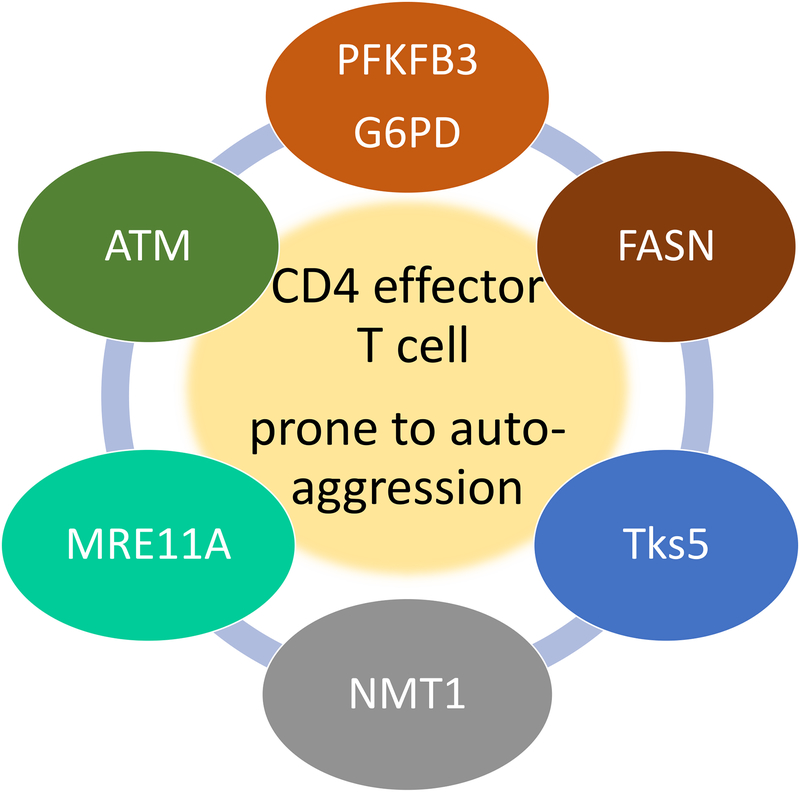
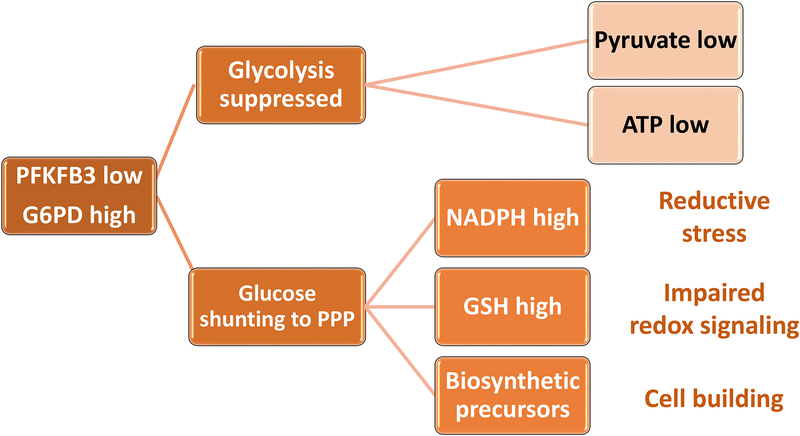
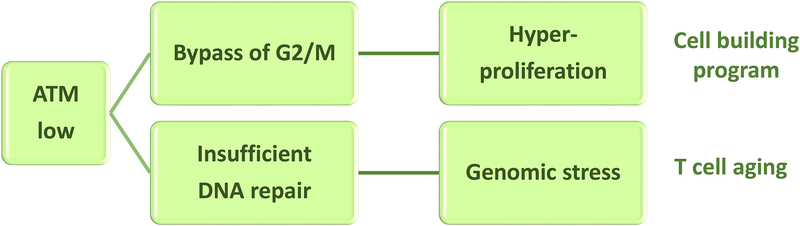
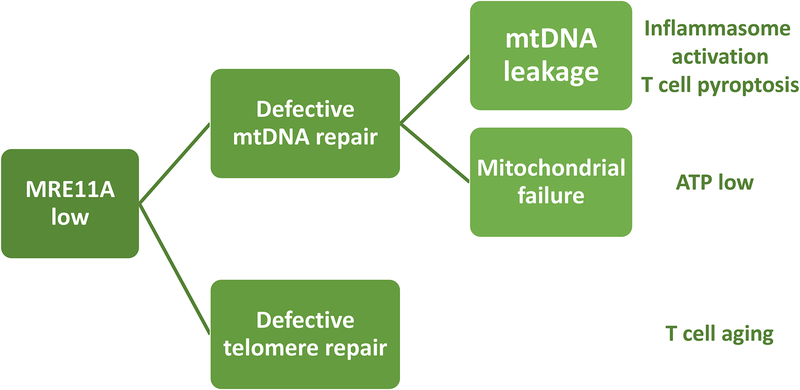
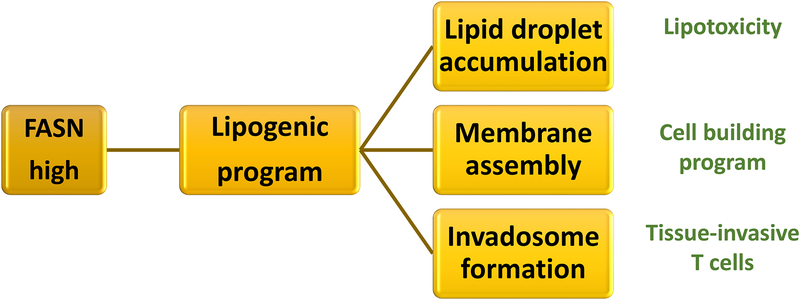
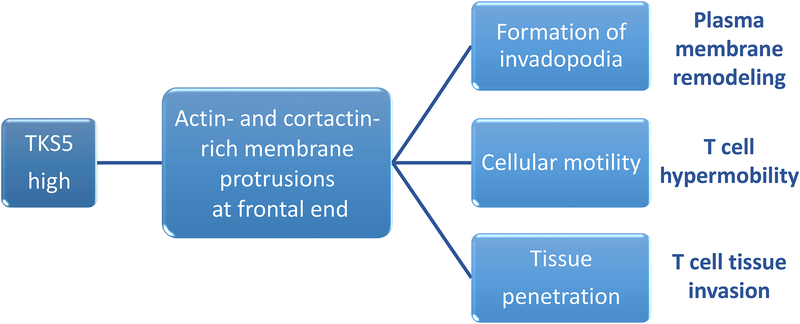
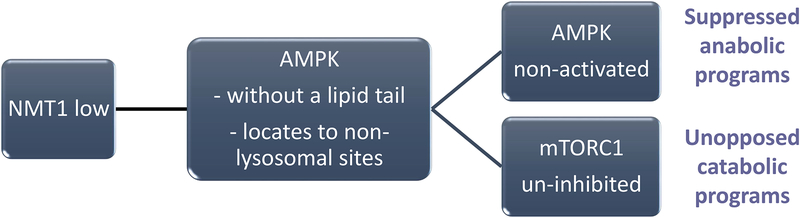
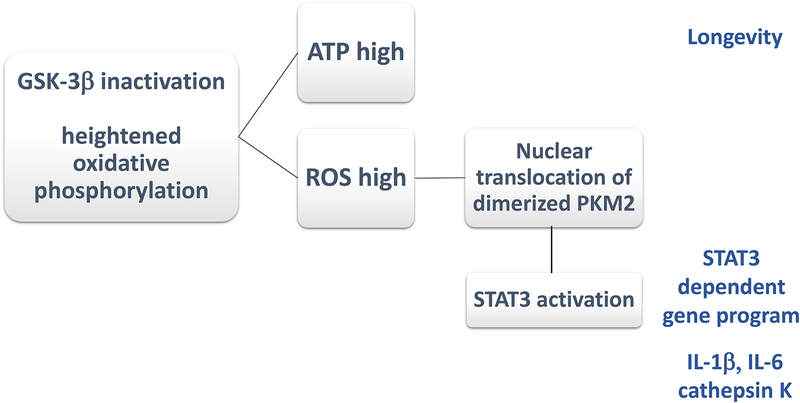
In rheumatoid arthritis (RA), breakdown of self-tolerance and onset of clinical disease are separated in time and space, supporting a multi-hit model in which emergence of autoreactive T cells is a pinnacle pathogenic event. Determining factors in T cell differentiation and survival include antigen recognition, but also the metabolic machinery that provides energy and biosynthetic molecules for cell building. Studies in patients with RA have yielded a disease-specific metabolic signature, which enables naive CD4 T cells to differentiate into pro-inflammatory helper T cells that are prone to invade into tissue and elicit inflammation through immunogenic cell death. A typifying property of RA CD4 T cells is the shunting of glucose away from glycolytic breakdown and mitochondrial processing toward the pentose phosphate pathway, favoring anabolic over catabolic reactions. Key defects have been localized to the mitochondria and the lysosome; including instability of mitochondrial DNA due to the lack of the DNA repair nuclease MRE11A and inefficient lysosomal tethering of AMPK due to deficiency of N-myristoyltransferase 1 (NMT1). The molecular taxonomy of the metabolically reprogrammed RA T cells includes glycolytic enzymes (glucose-6-phosphate dehydrogenase, phosphofructokinase), DNA repair molecules (MRE11A, ATM), regulators of protein trafficking (NMT1), and the membrane adapter protein TSK5. As the mechanisms determining abnormal T cell behavior in RA are unraveled, opportunities will emerge to interject autoimmune T cells by targeting their metabolic checkpoints.
Keywords: DNA damage; DNA repair; T cell; autoimmunity; cell cycle; glycolysis; macrophage; mitochondria; myristoylation; protein trafficking; rheumatoid arthritis; telomere.
© 2020 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
Figures
References
-
- Martins P, Fonseca JE. How to investigate: Pre-clinical rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2019;101438. - PubMed
-
- Kalinkovich A, Gabdulina G, Livshits G. Autoimmunity, inflammation, and dysbiosis mutually govern the transition from the preclinical to the clinical stage of rheumatoid arthritis. Immunol Res. 2018;66(6):696–709. - PubMed
-
- Ma WT, Chang C, Gershwin ME, Lian ZX. Development of autoantibodies precedes clinical manifestations of autoimmune diseases: A comprehensive review. J Autoimmun. 2017;8395–112. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
