

Novel KLHL26 variant associated with a familial case of Ebstein's anomaly and left ventricular noncompaction
- PMID: 31985165
- PMCID: PMC7196453
- DOI: 10.1002/mgg3.1152
Novel KLHL26 variant associated with a familial case of Ebstein's anomaly and left ventricular noncompaction
Abstract
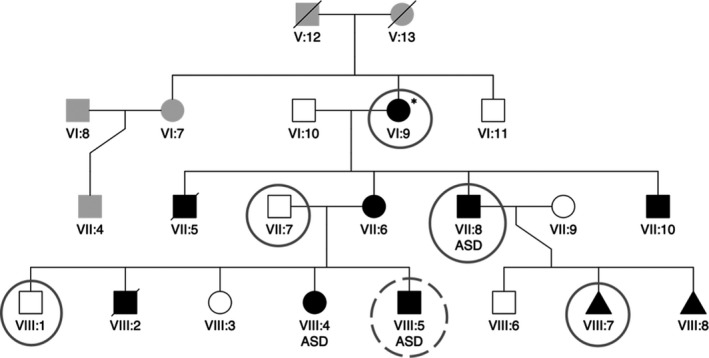
Background: Ebstein's anomaly (EA) is a rare congenital heart disease of the tricuspid valve and right ventricle. Patients with EA often manifest with left ventricular noncompaction (LVNC), a cardiomyopathy. Despite implication of cardiac sarcomere genes in some cases, very little is understood regarding the genetic etiology of EA/LVNC. Our study describes a multigenerational family with at least 10 of 17 members affected by EA/LVNC.
Methods: We performed echocardiography on all family members and conducted exome sequencing of six individuals. After identifying candidate variants using two different bioinformatic strategies, we confirmed segregation with phenotype using Sanger sequencing. We investigated structural implications of candidate variants using protein prediction models.
Results: Exome sequencing analysis of four affected and two unaffected members identified a novel, rare, and damaging coding variant in the Kelch-like family member 26 (KLHL26) gene located on chromosome 19 at position 237 of the protein (GRCh37). This variant region was confirmed by Sanger sequencing in the remaining family members. KLHL26 (c.709C > T p.R237C) segregates only with EA/LVNC-affected individuals (FBAT p < .05). Investigating structural implications of the candidate variant using protein prediction models suggested that the KLHL26 variant disrupts electrostatic interactions when binding to part of the ubiquitin proteasome, specifically Cullin3 (CUL3), a component of E3 ubiquitin ligase.
Conclusion: In this familial case of EA/LVNC, we have identified a candidate gene variant, KLHL26 (p.R237C), which may have an important role in ubiquitin-mediated protein degradation during cardiac development.
Keywords: Ebstein's anomaly; Kelch-like family member 26; congenital heart defects; left-ventricular noncompaction; ubiquitin proteasome.
© 2020 The Authors. Molecular Genetics & Genomic Medicine published by Wiley Periodicals, Inc.
Conflict of interest statement
A. Tomita‐Mitchell and M. E. Mitchell are cofounders of TAI Diagnostics (Milwaukee, WI), a biotechnology company involved in transplant diagnostics, and members of its scientific advisory board.
Figures


References
-
- Benson, D. W. , Silberbach, G. M. , Kavanaugh‐McHugh, A. , Cottrill, C. , Zhang, Y. , Riggs, S. , … Kugler, J. D. (1999). Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. Journal of Clinical Investigation, 104(11), 1567–1573. 10.1172/JCI8154 - DOI - PMC - PubMed
-
- Bettinelli, A. L. , Mulder, T. J. , Funke, B. H. , Lafferty, K. A. , Longo, S. A. , & Niyazov, D. M. (2013). Familial ebstein anomaly, left ventricular hypertrabeculation, and ventricular septal defect associated with a MYH7 mutation. American Journal of Medical Genetics. Part A, 161A(12), 3187–3190. 10.1002/ajmg.a.36182 - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
