

Deep learning models predict regulatory variants in pancreatic islets and refine type 2 diabetes association signals
- PMID: 31985400
- PMCID: PMC7007221
- DOI: 10.7554/eLife.51503
Deep learning models predict regulatory variants in pancreatic islets and refine type 2 diabetes association signals
Abstract
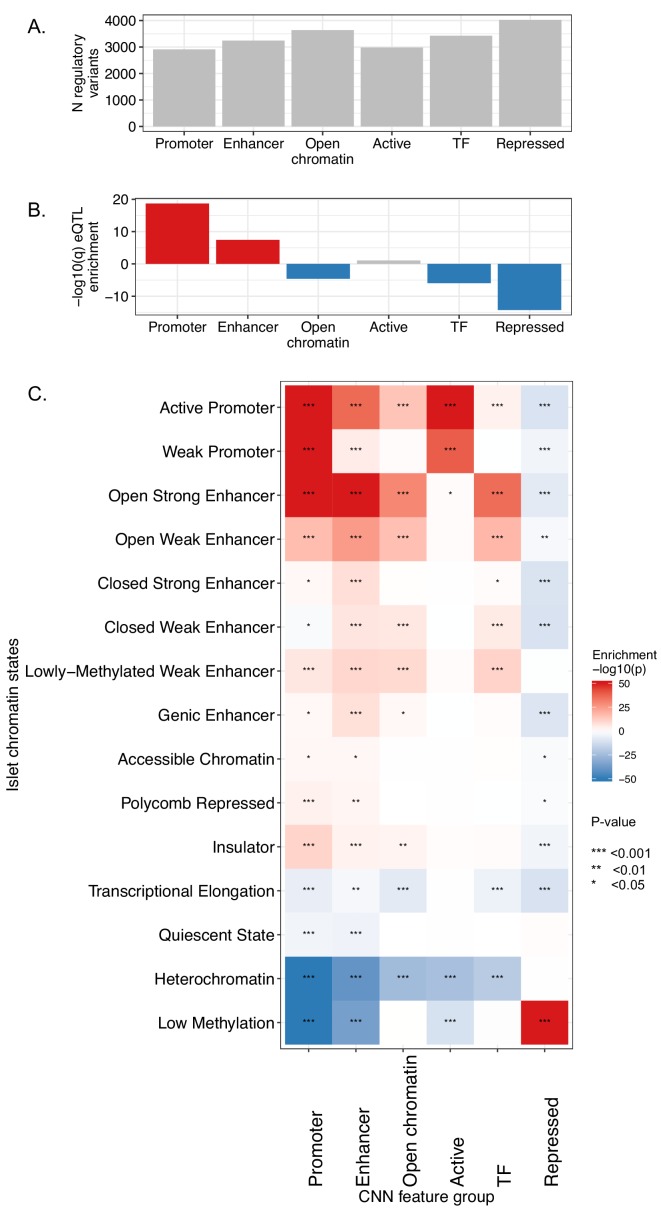
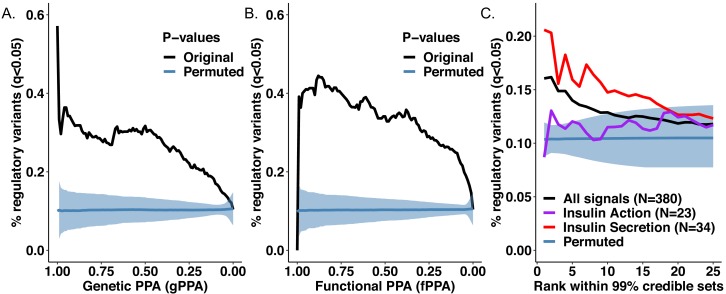
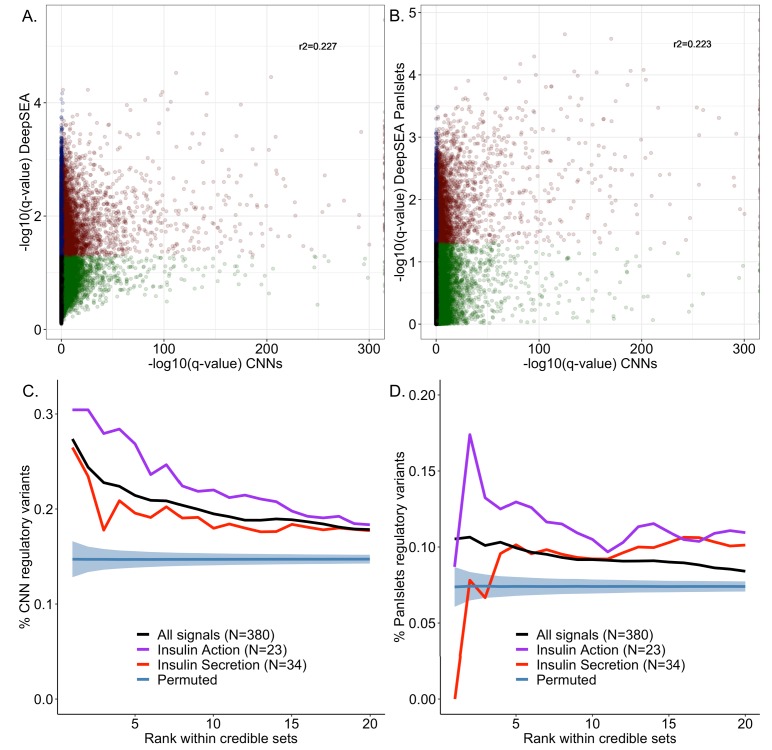
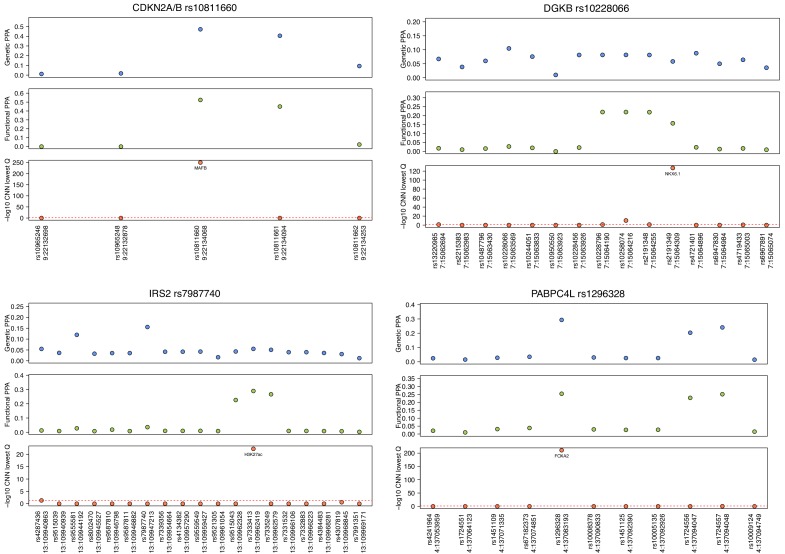
Genome-wide association analyses have uncovered multiple genomic regions associated with T2D, but identification of the causal variants at these remains a challenge. There is growing interest in the potential of deep learning models - which predict epigenome features from DNA sequence - to support inference concerning the regulatory effects of disease-associated variants. Here, we evaluate the advantages of training convolutional neural network (CNN) models on a broad set of epigenomic features collected in a single disease-relevant tissue - pancreatic islets in the case of type 2 diabetes (T2D) - as opposed to models trained on multiple human tissues. We report convergence of CNN-based metrics of regulatory function with conventional approaches to variant prioritization - genetic fine-mapping and regulatory annotation enrichment. We demonstrate that CNN-based analyses can refine association signals at T2D-associated loci and provide experimental validation for one such signal. We anticipate that these approaches will become routine in downstream analyses of GWAS.
Keywords: computational biology; convolutional neural networks; deep learning; epigenomics; fine-mapping; genetics; genomics; human; pancreatic islets; systems biology; type 2 diabetes.
© 2020, Wesolowska-Andersen et al.
Conflict of interest statement
AW, GZ, VN, FA, MT, JT, AM, AG No competing interests declared, MM Senior editor, eLife. MMcC has served on advisory panels for Pfizer, NovoNordisk and Zoe Global, has received honoraria from Merck, Pfizer, Novo Nordisk and Eli Lilly, and research funding from Abbvie, Astra Zeneca, Boehringer Ingelheim, Eli Lilly, Janssen, Merck, NovoNordisk, Pfizer, Roche, Sanofi Aventis, Servier, and Takeda. As of June 2019, MMcC is an employee of Genentech, and a holder of Roche stock.
Figures
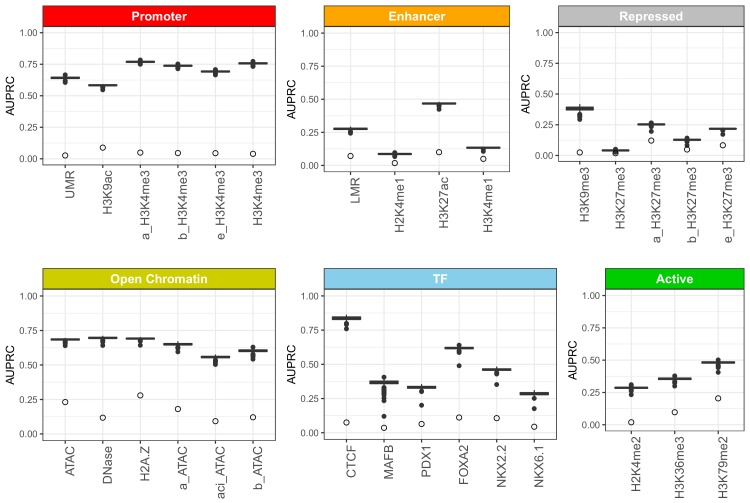
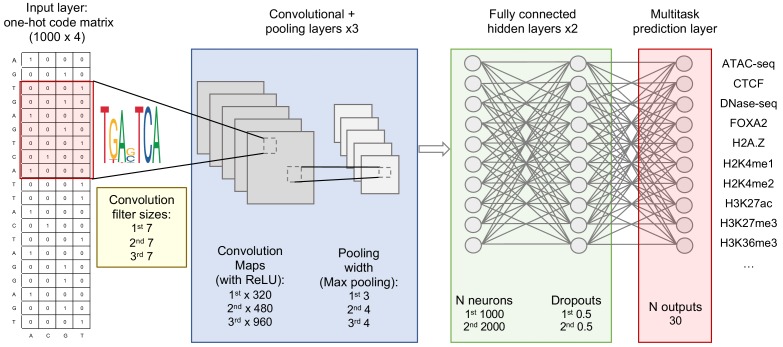
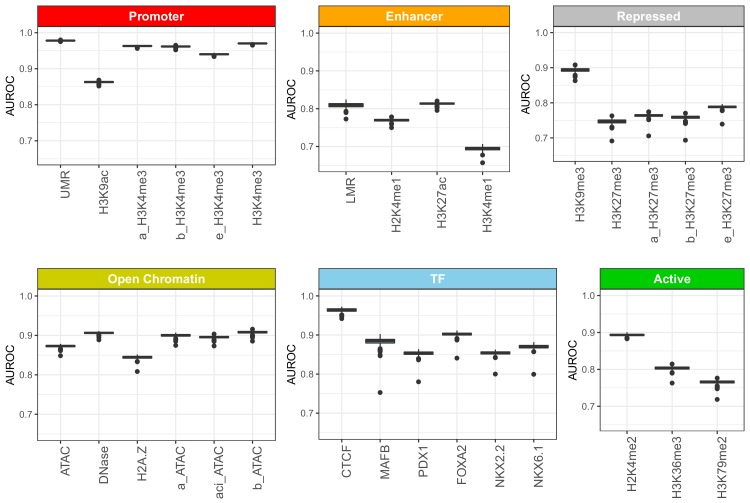
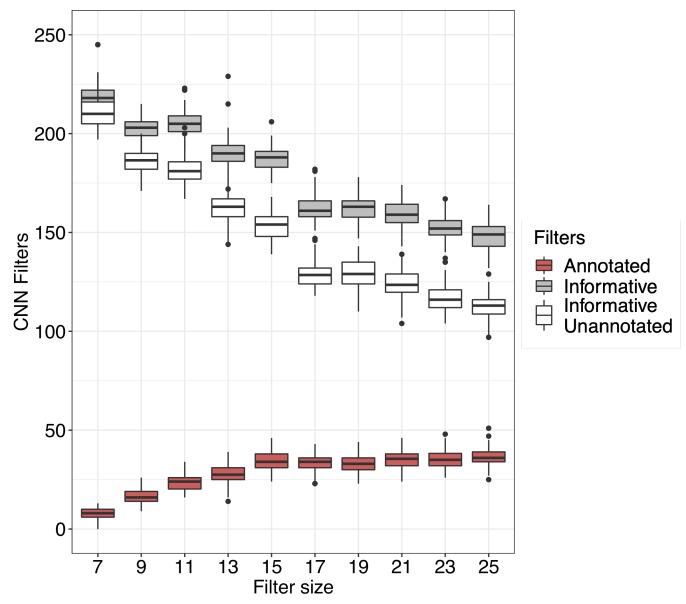
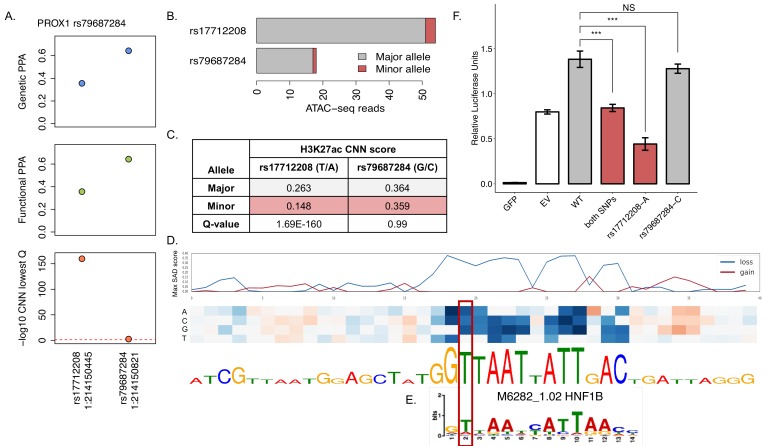
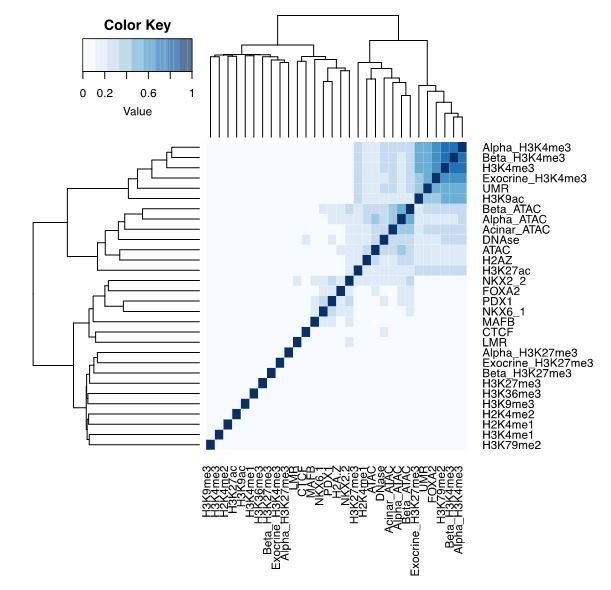
References
-
- Battle A, Brown CD, Engelhardt BE, Montgomery SB, GTEx Consortium. Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group. Statistical Methods groups—Analysis Working Group. Enhancing GTEx (eGTEx) groups. NIH Common Fund. NIH/NCI. NIH/NHGRI. NIH/NIMH. NIH/NIDA. Biospecimen Collection Source Site—NDRI. Biospecimen Collection Source Site—RPCI. Biospecimen Core Resource—VARI. Brain Bank Repository—University of Miami Brain Endowment Bank. Leidos Biomedical—Project Management. ELSI Study. Genome Browser Data Integration &Visualization—EBI. Genome Browser Data Integration &Visualization—UCSC Genomics Institute, University of California Santa Cruz. Lead analysts: Laboratory, Data Analysis &Coordinating Center (LDACC): NIH program management: Biospecimen collection: Pathology: eQTL manuscript working group: Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. - DOI - PMC - PubMed
-
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M, Lander ES, Mikkelsen TS, Thomson JA. The NIH roadmap epigenomics mapping consortium. Nature Biotechnology. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
