

Loss of Kat2a enhances transcriptional noise and depletes acute myeloid leukemia stem-like cells
- PMID: 31985402
- PMCID: PMC7039681
- DOI: 10.7554/eLife.51754
Loss of Kat2a enhances transcriptional noise and depletes acute myeloid leukemia stem-like cells
Abstract
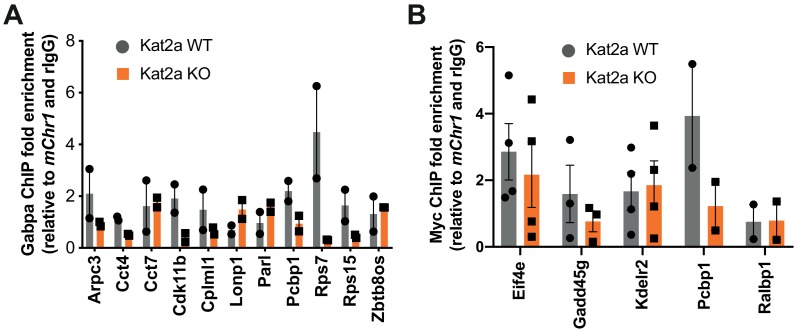
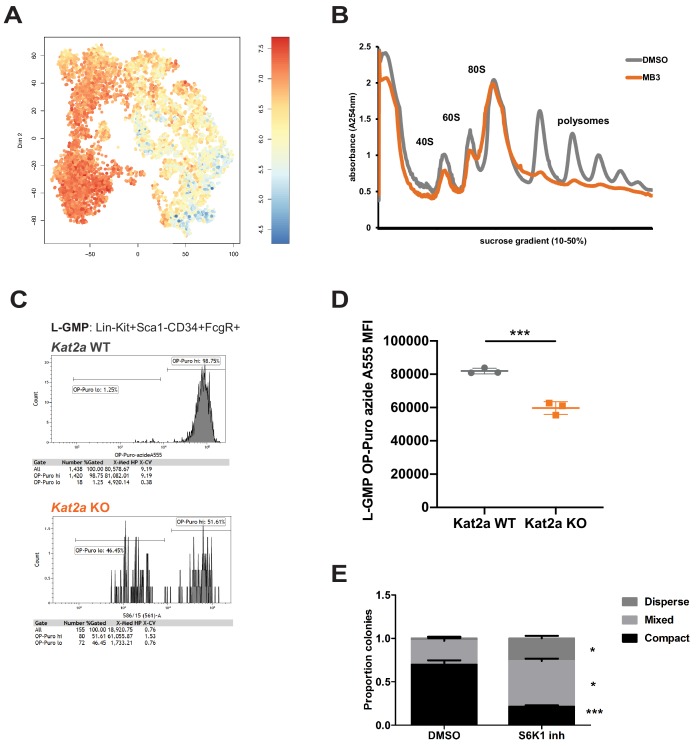
Acute Myeloid Leukemia (AML) is an aggressive hematological malignancy with abnormal progenitor self-renewal and defective white blood cell differentiation. Its pathogenesis comprises subversion of transcriptional regulation, through mutation and by hijacking normal chromatin regulation. Kat2a is a histone acetyltransferase central to promoter activity, that we recently associated with stability of pluripotency networks, and identified as a genetic vulnerability in AML. Through combined chromatin profiling and single-cell transcriptomics of a conditional knockout mouse, we demonstrate that Kat2a contributes to leukemia propagation through preservation of leukemia stem-like cells. Kat2a loss impacts transcription factor binding and reduces transcriptional burst frequency in a subset of gene promoters, generating enhanced variability of transcript levels. Destabilization of target programs shifts leukemia cell fate out of self-renewal into differentiation. We propose that control of transcriptional variability is central to leukemia stem-like cell propagation, and establish a paradigm exploitable in different tumors and distinct stages of cancer evolution.
Keywords: Kat2a; Single-cell RNA seq; acute myeloid leukaemia; cancer biology; chromosomes; epigenetics; gene expression; mouse; transcriptional bursting; transcriptional heterogeneity.
Plain language summary
Less than 30% of patients with acute myeloid leukaemia – an aggressive cancer of the white blood cells – survive five years post-diagnosis. This disease disrupts the maturation of white blood cells, resulting in the accumulation of immature cells that multiply and survive but are incapable of completing their maturation process. Amongst these, a group of cancer cells known as leukemic stem cells is responsible for continually replenishing the leukaemia, thus perpetuating its growth. Cancers develop when cells in the body acquire changes or mutations to their genetic makeup. The mutations that lead to acute myeloid leukaemia often affect the activity of genes known as epigenetic regulators. These genes regulate which proteins and other molecules cells make by controlling the way in which cells ‘read’ their genetic instructions. The epigenetic regulator Kat2a is thought to ‘tune’ the frequency at which cells read their genetic instructions. This tuning mechanism decreases random fluctuations in the execution of the instructions cells receive to make proteins and other molecules. In turn, this helps to ensure that individual cells of the same type behave in a similar way, for example by keeping leukaemia cells in an immature state. Here, Domingues, Kulkarni et al. investigated whether interfering with Kat2a can make acute myeloid leukaemia less aggressive by allowing the immature white blood cells to mature. Domingues, Kulkarni et al. genetically engineered mice to remove Kat2a from blood cells on demand and then inserted a mutation that causes acute myeloid leukaemia. The experiments showed that the loss of Kat2a delayed the development of leukaemia in the mice and progressively depleted leukaemia stem cells, causing the disease to become less aggressive. The results also showed that loss of Kat2a caused more fluctuations in how the white blood cells read their genetic code, which resulted in more variability in the molecules they produced and increased the tendency of the cells to mature. These findings establish that loss of Kat2a causes leukaemia stem cells to mature and stop multiplying by untuning the frequency at which the cells read their genetic instructions. In the future, it may be possible to develop drugs that target human KAT2A to treat acute myeloid leukaemia.
© 2020, Domingues et al.
Conflict of interest statement
AD, RK, GG, SG, LV, LA, EF, MK, RA, AJ, ST, KZ, BH, SP, CP No competing interests declared
Figures
Similar articles
-
Buffering noise: KAT2A modular contributions to stabilization of transcription and cell identity in cancer and development.Exp Hematol. 2021 Jan;93:25-37. doi: 10.1016/j.exphem.2020.10.003. Epub 2020 Oct 22. Exp Hematol. 2021. PMID: 33223444
-
Contributions of transcriptional noise to leukaemia evolution: KAT2A as a case-study.Philos Trans R Soc Lond B Biol Sci. 2024 Apr 22;379(1900):20230052. doi: 10.1098/rstb.2023.0052. Epub 2024 Mar 4. Philos Trans R Soc Lond B Biol Sci. 2024. PMID: 38432321 Free PMC article. Review.
-
Convergent epigenetic evolution drives relapse in acute myeloid leukemia.Elife. 2024 Apr 22;13:e93019. doi: 10.7554/eLife.93019. Elife. 2024. PMID: 38647535 Free PMC article.
-
Histone Acetyltransferase KAT2A Stabilizes Pluripotency with Control of Transcriptional Heterogeneity.Stem Cells. 2018 Dec;36(12):1828-1838. doi: 10.1002/stem.2919. Epub 2018 Oct 17. Stem Cells. 2018. PMID: 30270482 Free PMC article.
-
Regulating the leukaemia stem cell.Best Pract Res Clin Haematol. 2009 Dec;22(4):483-7. doi: 10.1016/j.beha.2009.08.005. Best Pract Res Clin Haematol. 2009. PMID: 19959097 Free PMC article. Review.
Cited by
-
An evolutionary approach to clonally complex hematologic disorders.Blood Cancer Discov. 2021 May;2(3):201-215. doi: 10.1158/2643-3230.BCD-20-0219. Epub 2021 Apr 15. Blood Cancer Discov. 2021. PMID: 34027415 Free PMC article. Review.
-
Transcriptional variability accelerates preleukemia by cell diversification and perturbation of protein synthesis.Sci Adv. 2022 Aug 5;8(31):eabn4886. doi: 10.1126/sciadv.abn4886. Epub 2022 Aug 3. Sci Adv. 2022. PMID: 35921412 Free PMC article.
-
Bidirectional interplay between metabolism and epigenetics in hematopoietic stem cells and leukemia.EMBO J. 2023 Dec 11;42(24):e112348. doi: 10.15252/embj.2022112348. Epub 2023 Nov 27. EMBO J. 2023. PMID: 38010205 Free PMC article. Review.
-
KAT2A complexes ATAC and SAGA play unique roles in cell maintenance and identity in hematopoiesis and leukemia.Blood Adv. 2022 Jan 11;6(1):165-180. doi: 10.1182/bloodadvances.2020002842. Blood Adv. 2022. PMID: 34654054 Free PMC article.
-
Co-Formulation of Amphiphilic Cationic and Anionic Cyclodextrins Forming Nanoparticles for siRNA Delivery in the Treatment of Acute Myeloid Leukaemia.Int J Mol Sci. 2022 Aug 29;23(17):9791. doi: 10.3390/ijms23179791. Int J Mol Sci. 2022. PMID: 36077202 Free PMC article.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
