

Mechanisms of Metabolic Acidosis-Induced Kidney Injury in Chronic Kidney Disease
- PMID: 31988269
- PMCID: PMC7062220
- DOI: 10.1681/ASN.2019070677
Mechanisms of Metabolic Acidosis-Induced Kidney Injury in Chronic Kidney Disease
Abstract
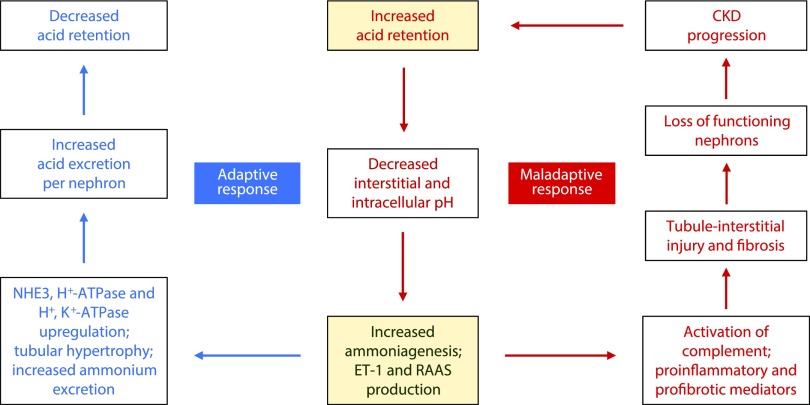
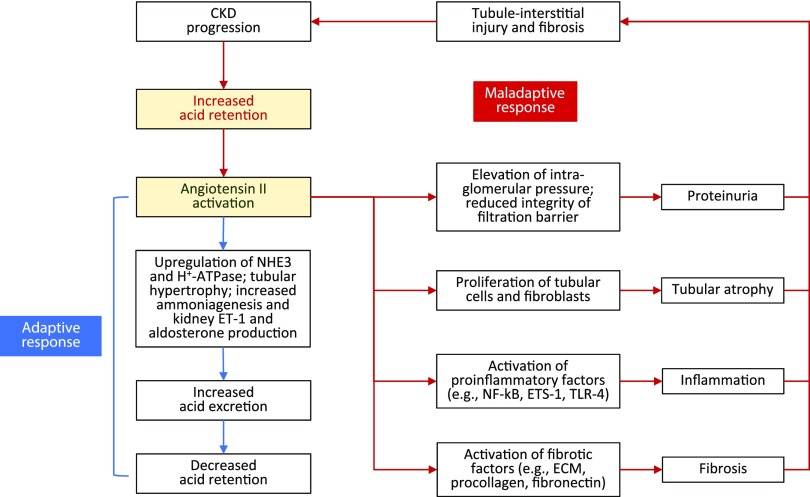
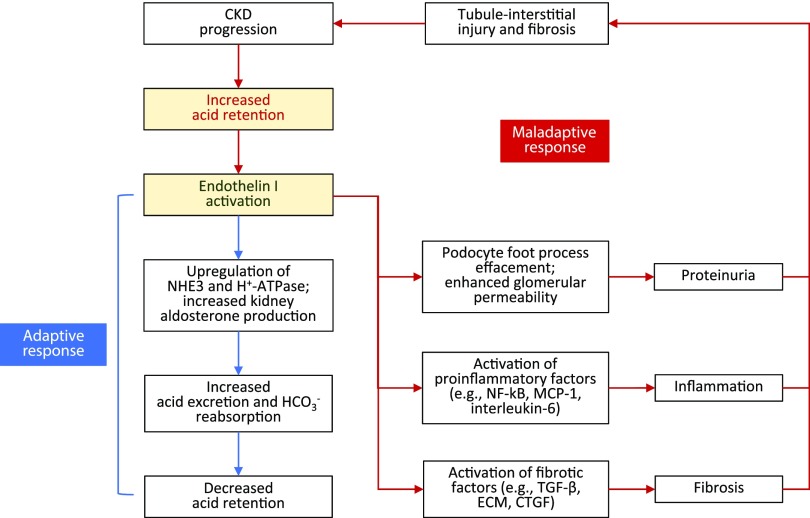
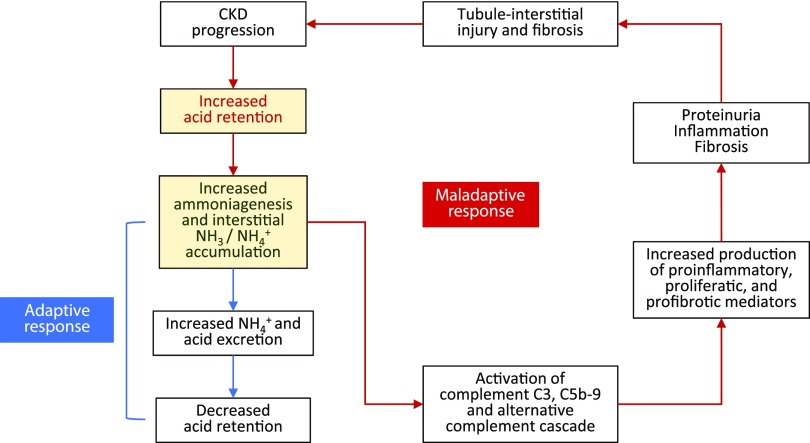
Retrospective analyses and single-center prospective studies identify chronic metabolic acidosis as an independent and modifiable risk factor for progression of CKD. In patients with CKD, untreated chronic metabolic acidosis often leads to an accelerated reduction in GFR. Mechanisms responsible for this reduction include adaptive responses that increase acid excretion but lead to a decline in kidney function. Metabolic acidosis in CKD stimulates production of intrakidney paracrine hormones including angiotensin II, aldosterone, and endothelin-1 (ET-1) that mediate the immediate benefit of increased kidney acid excretion, but their chronic upregulation promotes inflammation and fibrosis. Chronic metabolic acidosis also stimulates ammoniagenesis that increases acid excretion but also leads to ammonia-induced complement activation and deposition of C3 and C5b-9 that can cause tubule-interstitial damage, further worsening disease progression. These effects, along with acid accumulation in kidney tissue, combine to accelerate progression of kidney disease. Treatment of chronic metabolic acidosis attenuates these adaptive responses; reduces levels of angiotensin II, aldosterone, and ET-1; reduces ammoniagenesis; and diminishes inflammation and fibrosis that may lead to slowing of CKD progression.
Keywords: aldosterone; angiotensin; chronic kidney disease; chronic metabolic acidosis; endothelin; progression of chronic renal failure.
Copyright © 2020 by the American Society of Nephrology.
Figures
References
-
- Kraut JA, Madias NE: Metabolic acidosis of CKD: An update. Am J Kidney Dis 67: 307–317, 2016. - PubMed
-
- Alpern RJ, Sakhaee K: The clinical spectrum of chronic metabolic acidosis: Homeostatic mechanisms produce significant morbidity. Am J Kidney Dis 29: 291–302, 1997. - PubMed
-
- Kraut JA, Madias NE: Adverse effects of the metabolic acidosis of chronic kidney disease. Adv Chronic Kidney Dis 24: 289–297, 2017. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
