

Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy
- PMID: 31988462
- PMCID: PMC7212064
- DOI: 10.1038/s41591-019-0738-2
Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy
Abstract
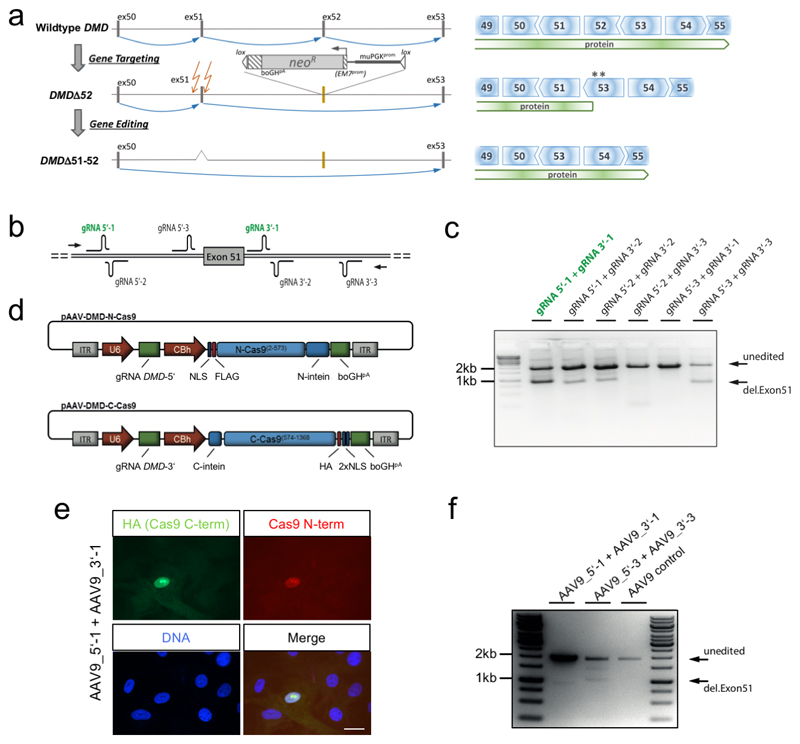
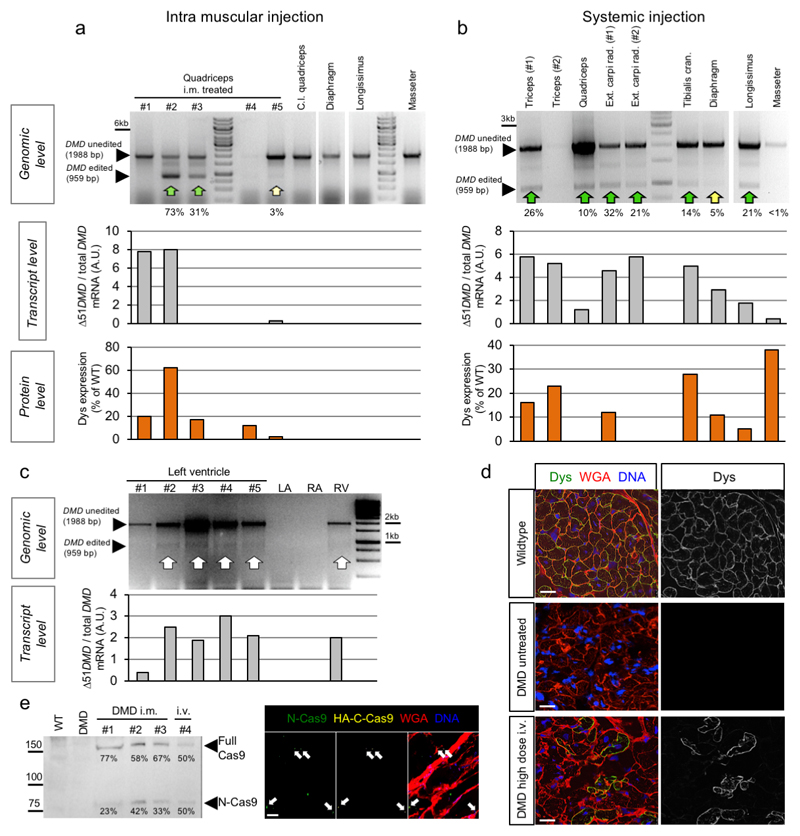
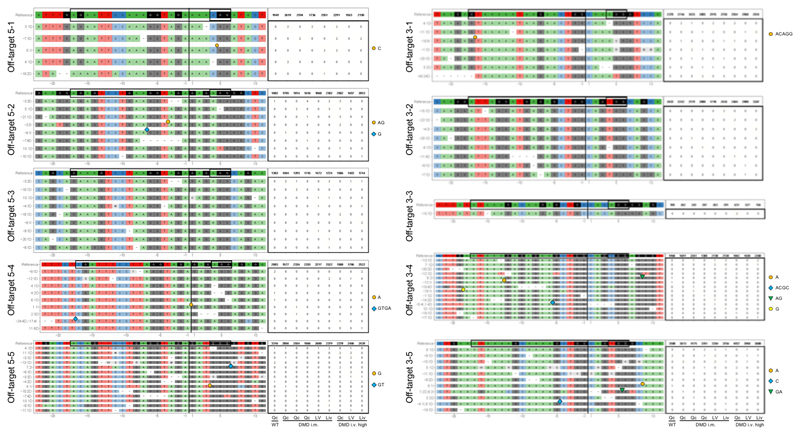
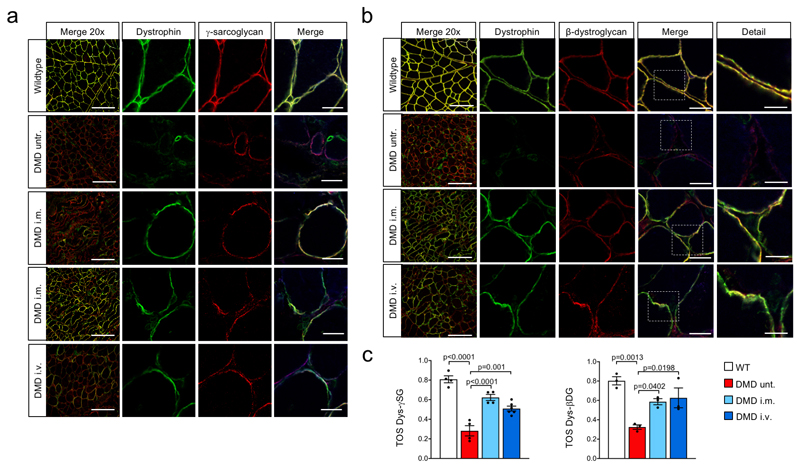
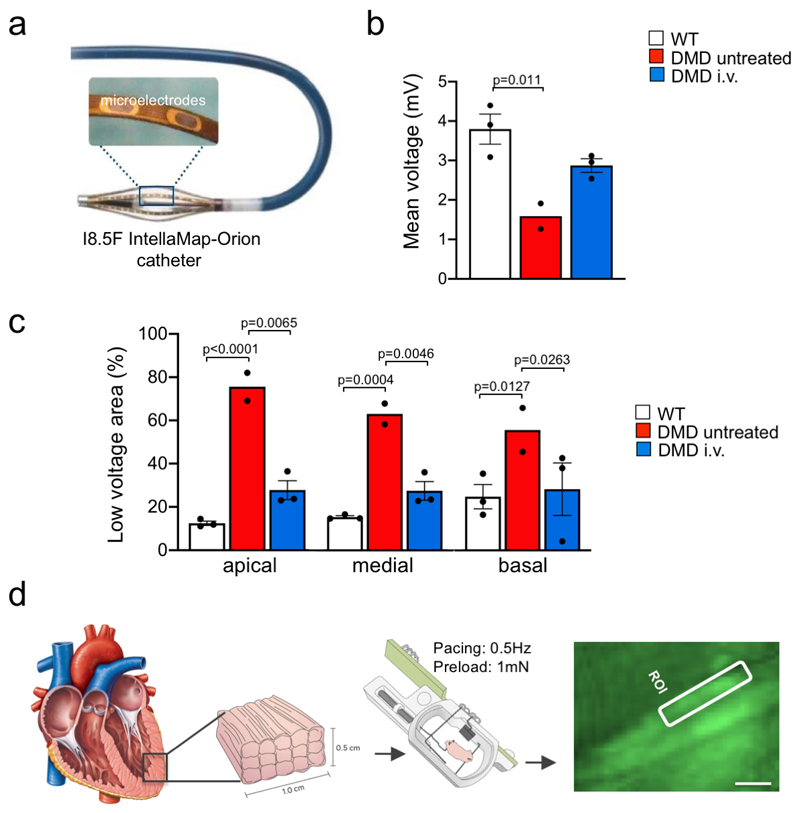
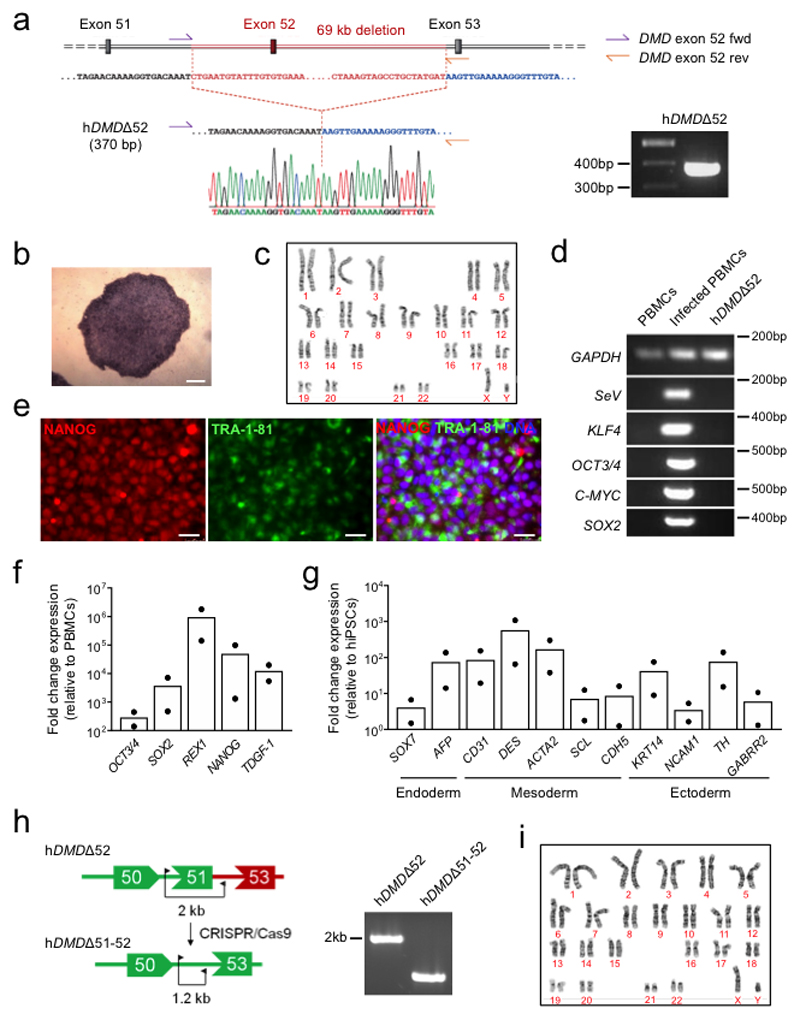
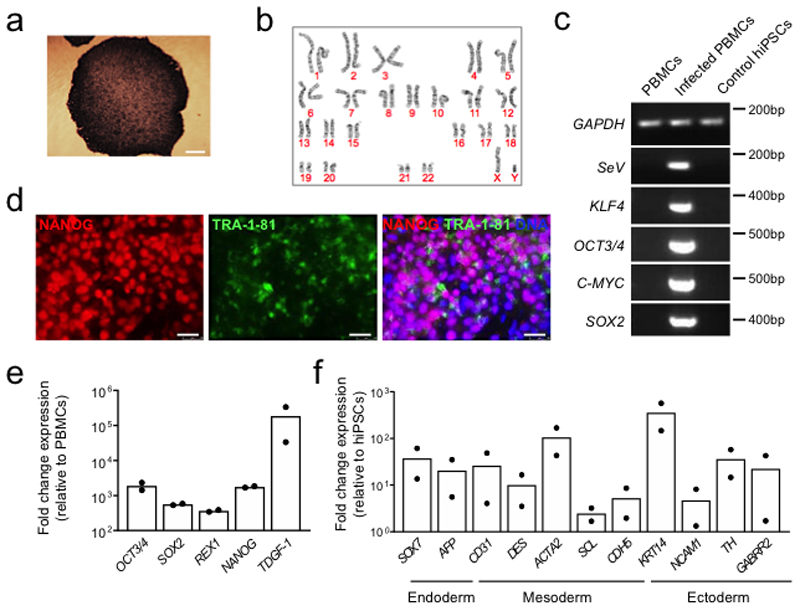
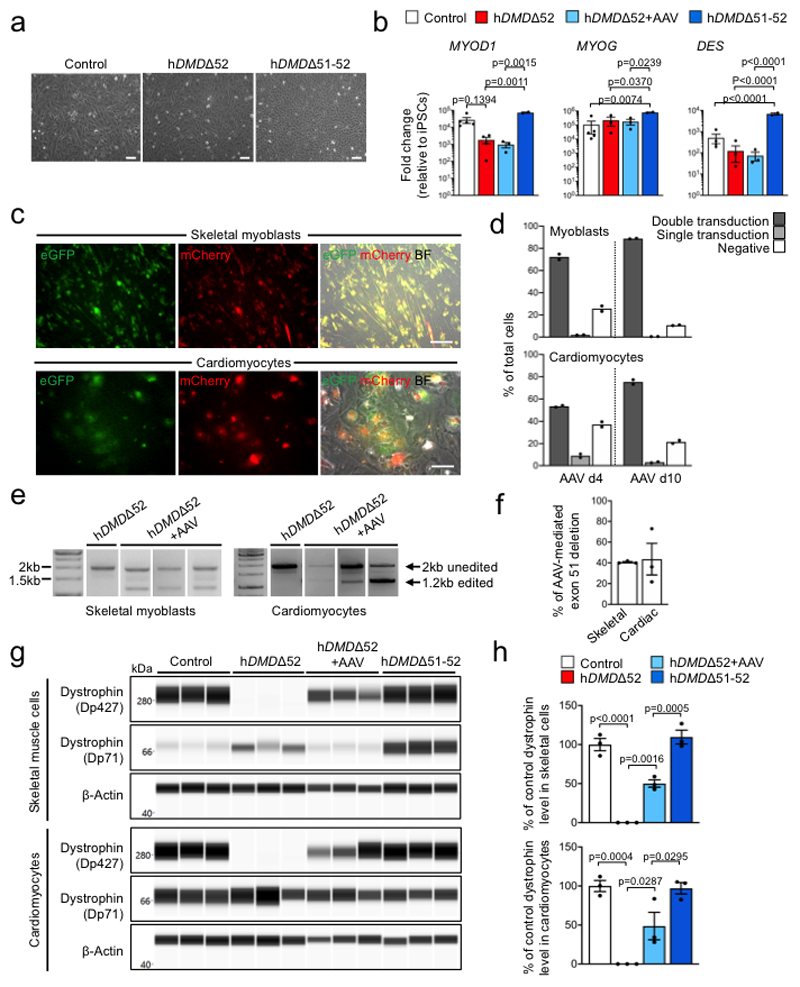
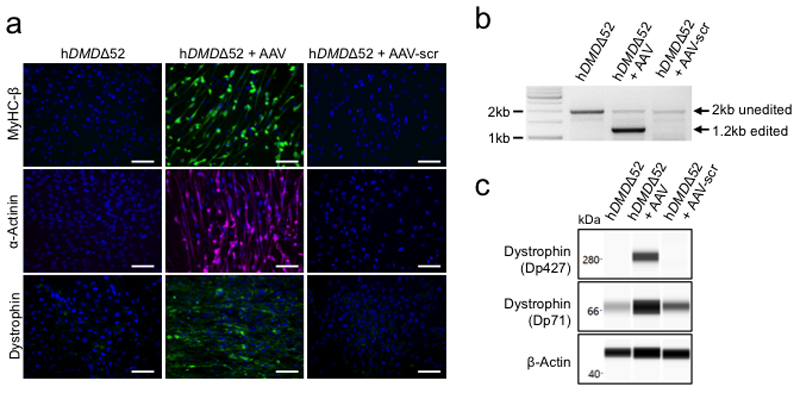
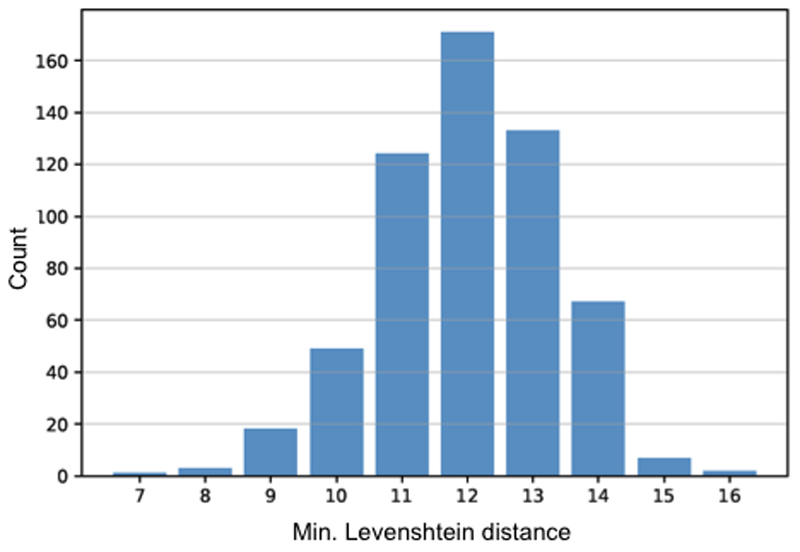
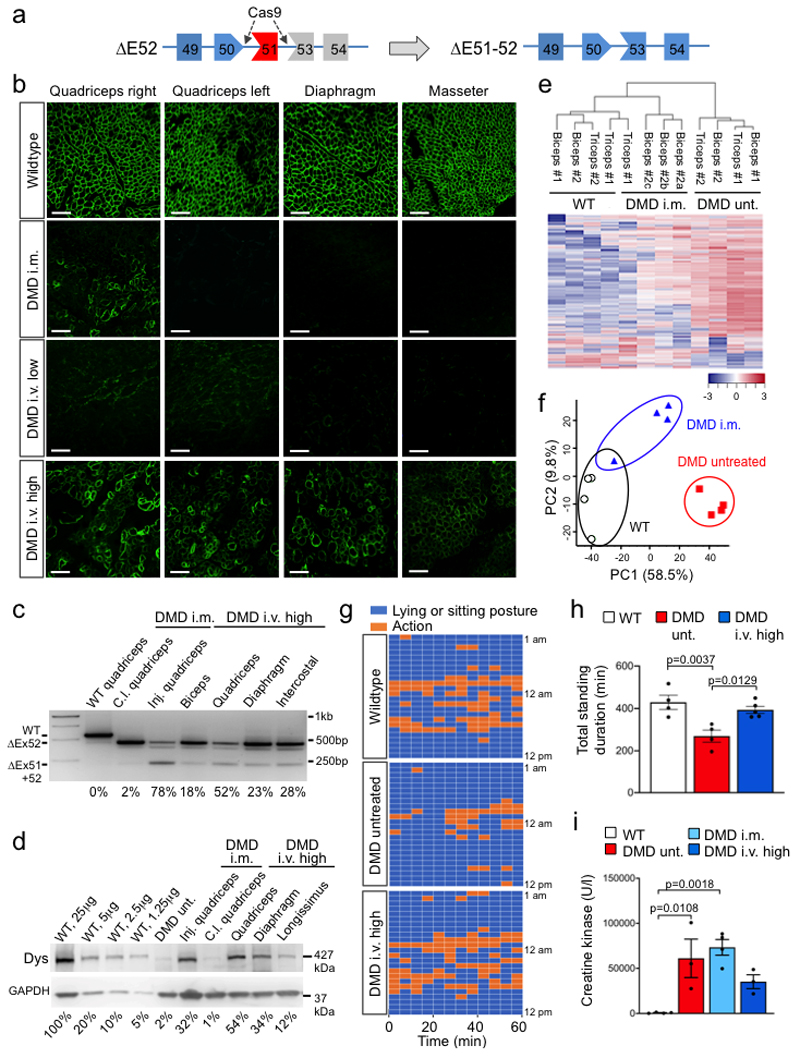
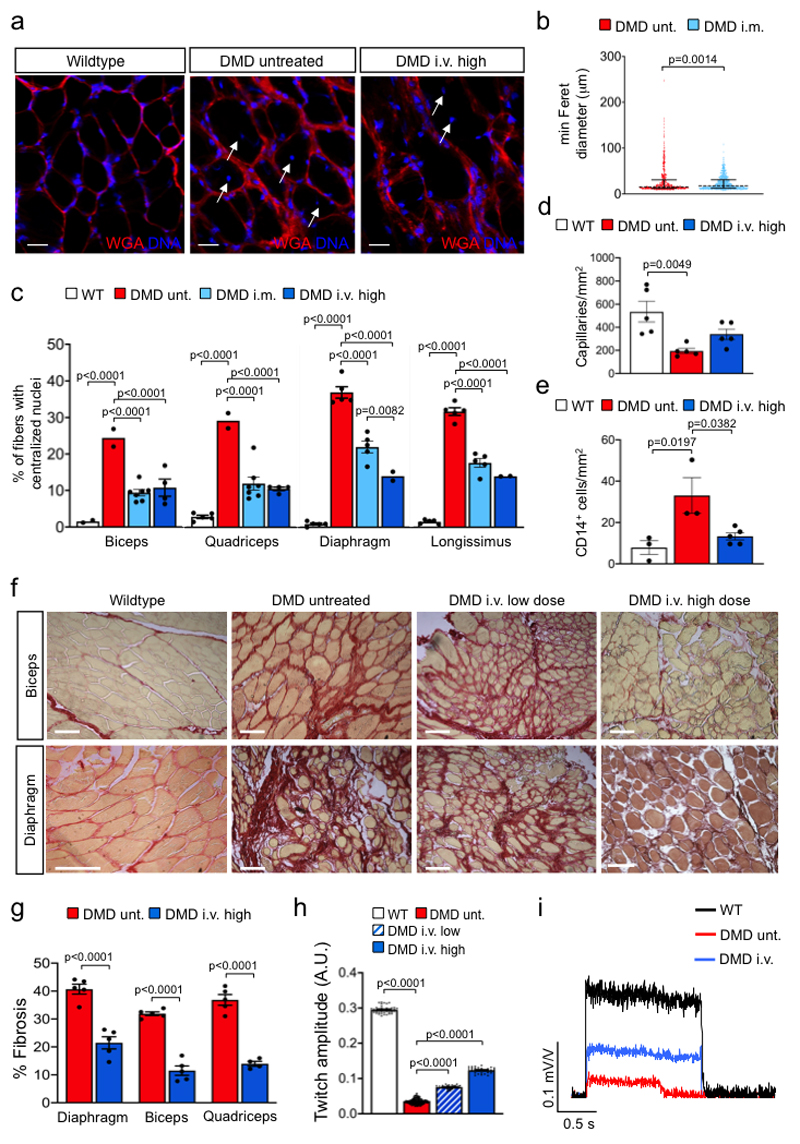
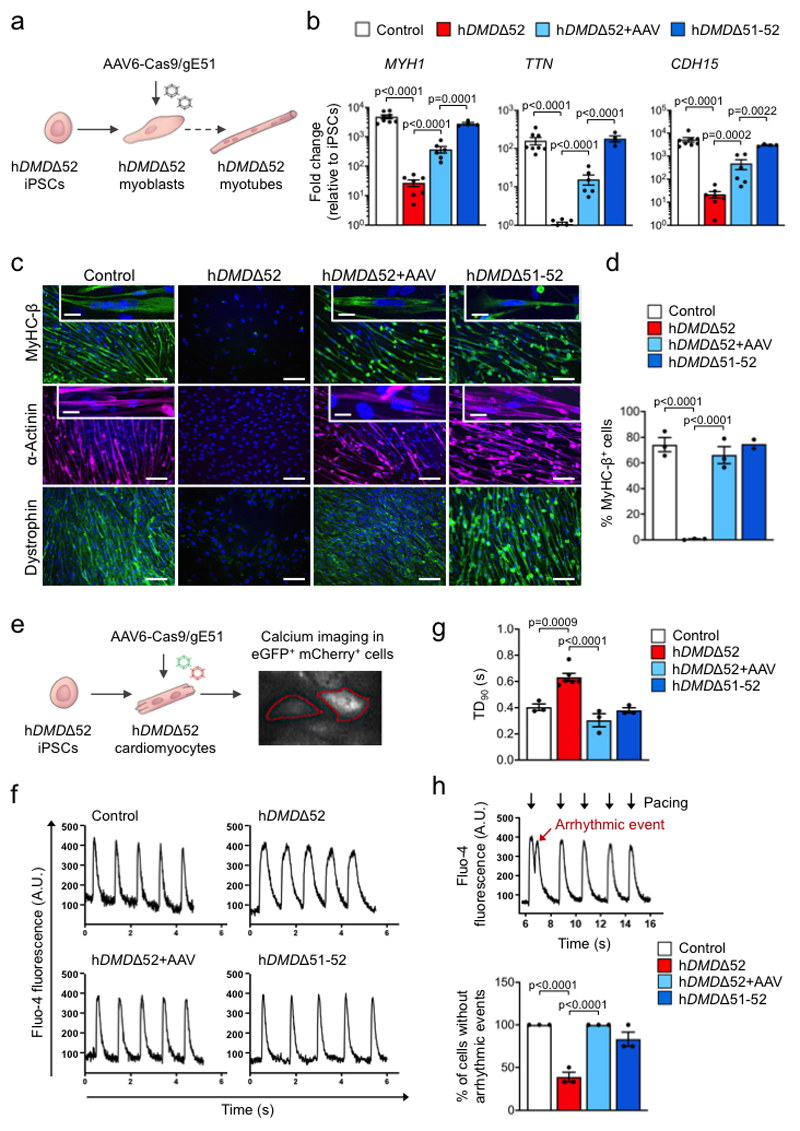
Frameshift mutations in the DMD gene, encoding dystrophin, cause Duchenne muscular dystrophy (DMD), leading to terminal muscle and heart failure in patients. Somatic gene editing by sequence-specific nucleases offers new options for restoring the DMD reading frame, resulting in expression of a shortened but largely functional dystrophin protein. Here, we validated this approach in a pig model of DMD lacking exon 52 of DMD (DMDΔ52), as well as in a corresponding patient-derived induced pluripotent stem cell model. In DMDΔ52 pigs1, intramuscular injection of adeno-associated viral vectors of serotype 9 carrying an intein-split Cas9 (ref. 2) and a pair of guide RNAs targeting sequences flanking exon 51 (AAV9-Cas9-gE51) induced expression of a shortened dystrophin (DMDΔ51-52) and improved skeletal muscle function. Moreover, systemic application of AAV9-Cas9-gE51 led to widespread dystrophin expression in muscle, including diaphragm and heart, prolonging survival and reducing arrhythmogenic vulnerability. Similarly, in induced pluripotent stem cell-derived myoblasts and cardiomyocytes of a patient lacking DMDΔ52, AAV6-Cas9-g51-mediated excision of exon 51 restored dystrophin expression and amelioreate skeletal myotube formation as well as abnormal cardiomyocyte Ca2+ handling and arrhythmogenic susceptibility. The ability of Cas9-mediated exon excision to improve DMD pathology in these translational models paves the way for new treatment approaches in patients with this devastating disease.
Conflict of interest statement
C.K. and W.W. have filed a patent for G2-AAV9-Cas9-gE51, which is covered by the results of Figures 1-3. All other authors have no competing interest.
Figures

Comment in
-
New developments in gene editing for Duchenne muscular dystrophy.Nat Rev Cardiol. 2020 Apr;17(4):200-201. doi: 10.1038/s41569-020-0350-7. Nat Rev Cardiol. 2020. PMID: 32047325 No abstract available.
References
-
- Klymiuk N, et al. Dystrophin-deficient pigs provide new insights into the hierarchy of physiological derangements of dystrophic muscle. Human Molecular Genetics. 2013;22:4368–4382. - PubMed
-
- Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle & nerve. 2006;34:135–144. - PubMed
-
- Moser H. Duchenne muscular dystrophy: pathogenetic aspects and genetic prevention. Human genetics. 1984;66:17–40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
