

Prader-Willi-Like Phenotype Caused by an Atypical 15q11.2 Microdeletion
- PMID: 31991769
- PMCID: PMC7073628
- DOI: 10.3390/genes11020128
Prader-Willi-Like Phenotype Caused by an Atypical 15q11.2 Microdeletion
Abstract
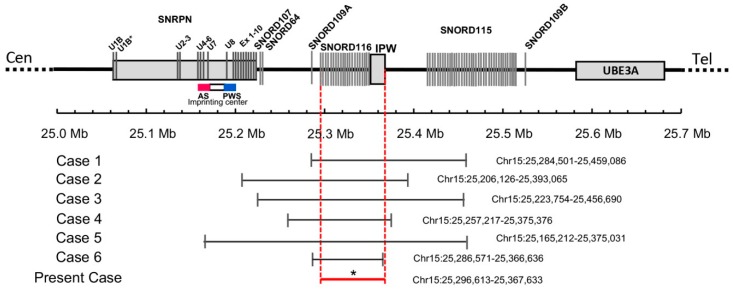
We report a 17-year-old boy who met most of the major Prader-Willi syndrome (PWS) diagnostic criteria, including infantile hypotonia and poor feeding followed by hyperphagia, early-onset morbid obesity, delayed development, and characteristic facial features. However, unlike many children with PWS, he had spontaneous onset of puberty and reached a tall adult stature without growth hormone replacement therapy. A phenotype-driven genetic analysis using exome sequencing identified a heterozygous microdeletion of 71 kb in size at chr15:25,296,613-25,367,633, genome build hg 19. This deletion does not affect the SNURF-SNRPN locus, but results in the loss of several of the PWS-associated non-coding RNA species, including the SNORD116 cluster. We compared with six previous reports of patients with PWS who carried small atypical deletions encompassing the snoRNA SNORD116 cluster. These patients share similar core symptoms of PWS while displaying some atypical features, suggesting that other genes in the region may make lesser phenotypic contributions. Altogether, these rare cases provide convincing evidence that loss of the paternal copy of the SNORD116 snoRNA is sufficient to cause most of the major clinical features of PWS.
Keywords: 15q11.2; Prader–Willi; SNORD116; atypical microdeletion.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
