

Unlocking the transcriptomic potential of formalin-fixed paraffin embedded clinical tissues: comparison of gene expression profiling approaches
- PMID: 31992186
- PMCID: PMC6988223
- DOI: 10.1186/s12859-020-3365-5
Unlocking the transcriptomic potential of formalin-fixed paraffin embedded clinical tissues: comparison of gene expression profiling approaches
Abstract
Background: High-throughput transcriptomics has matured into a very well established and widely utilised research tool over the last two decades. Clinical datasets generated on a range of different platforms continue to be deposited in public repositories provide an ever-growing, valuable resource for reanalysis. Cost and tissue availability normally preclude processing samples across multiple technologies, making it challenging to directly evaluate performance and whether data from different platforms can be reliably compared or integrated.
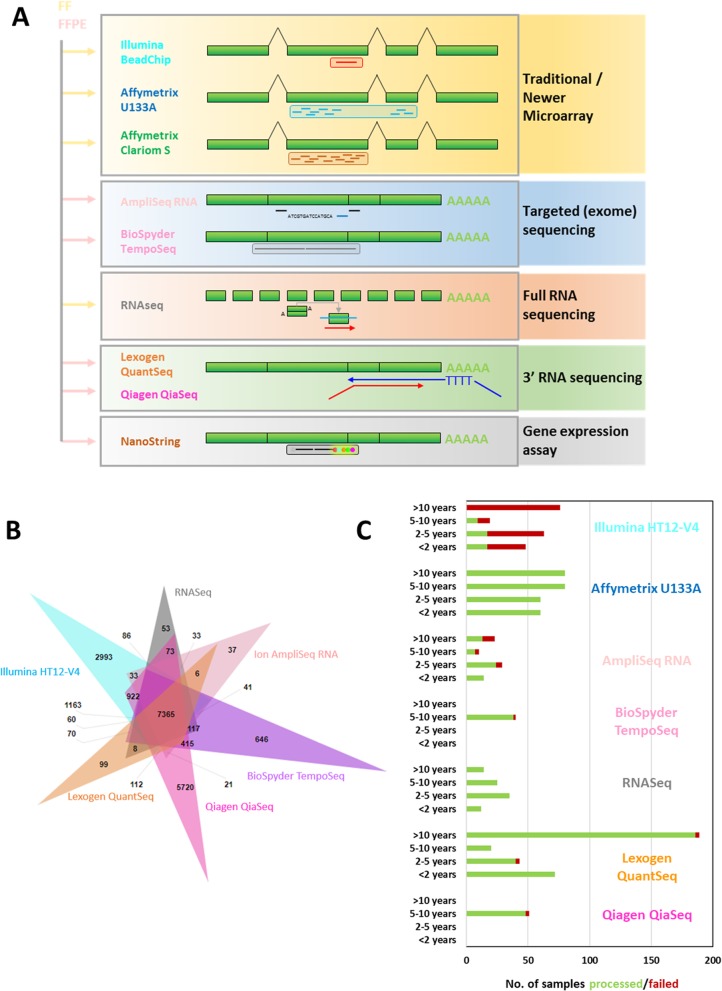
Methods: This study describes our experiences of nine new and established mRNA profiling techniques including Lexogen QuantSeq, Qiagen QiaSeq, BioSpyder TempO-Seq, Ion AmpliSeq, Nanostring, Affymetrix Clariom S or U133A, Illumina BeadChip and RNA-seq of formalin-fixed paraffin embedded (FFPE) and fresh frozen (FF) sequential patient-matched breast tumour samples.
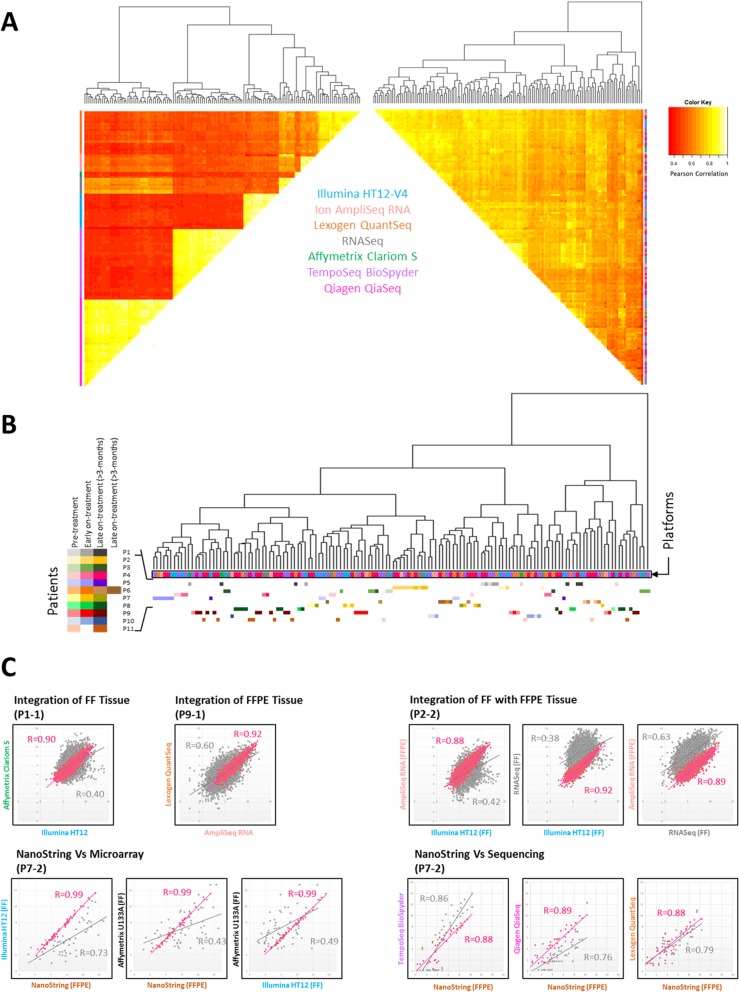
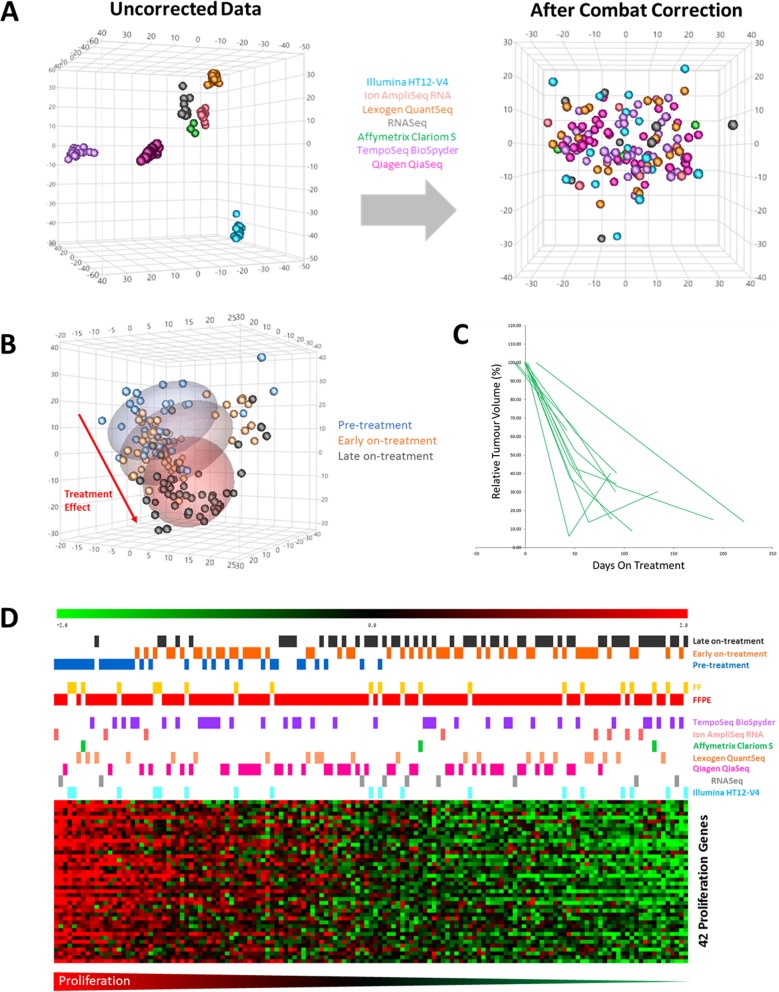
Results: The number of genes represented and reliability varied between the platforms, but overall all methods provided data which were largely comparable. Crucially we found that it is possible to integrate data for combined analyses across FFPE/FF and platforms using established batch correction methods as required to increase cohort sizes. However, some platforms appear to be better suited to FFPE samples, particularly archival material.
Conclusions: Overall, we illustrate that technology selection is a balance between required resolution, sample quality, availability and cost.
Keywords: FFPE; Fresh-frozen; Gene expression; Microarray; Sequencing; Transcriptomics.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
