

QuantTB - a method to classify mixed Mycobacterium tuberculosis infections within whole genome sequencing data
- PMID: 31992201
- PMCID: PMC6986090
- DOI: 10.1186/s12864-020-6486-3
QuantTB - a method to classify mixed Mycobacterium tuberculosis infections within whole genome sequencing data
Abstract
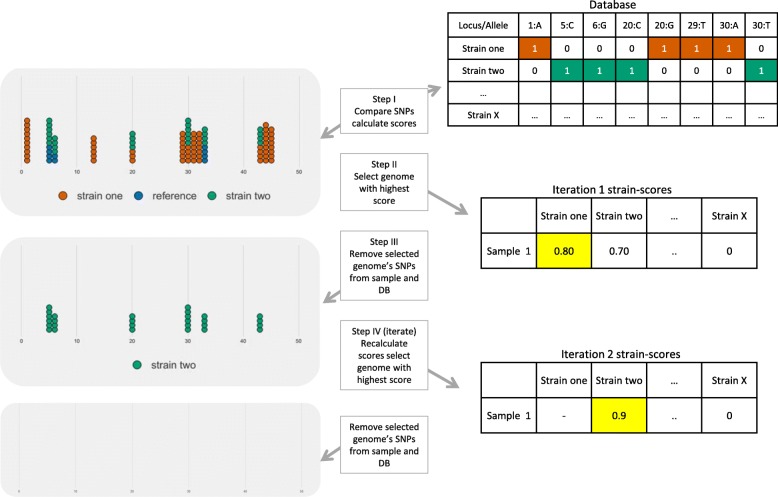
Background: Mixed infections of Mycobacterium tuberculosis and antibiotic heteroresistance continue to complicate tuberculosis (TB) diagnosis and treatment. Detection of mixed infections has been limited to molecular genotyping techniques, which lack the sensitivity and resolution to accurately estimate the multiplicity of TB infections. In contrast, whole genome sequencing offers sensitive views of the genetic differences between strains of M. tuberculosis within a sample. Although metagenomic tools exist to classify strains in a metagenomic sample, most tools have been developed for more divergent species, and therefore cannot provide the sensitivity required to disentangle strains within closely related bacterial species such as M. tuberculosis. Here we present QuantTB, a method to identify and quantify individual M. tuberculosis strains in whole genome sequencing data. QuantTB uses SNP markers to determine the combination of strains that best explain the allelic variation observed in a sample. QuantTB outputs a list of identified strains, their corresponding relative abundances, and a list of drugs for which resistance-conferring mutations (or heteroresistance) have been predicted within the sample.
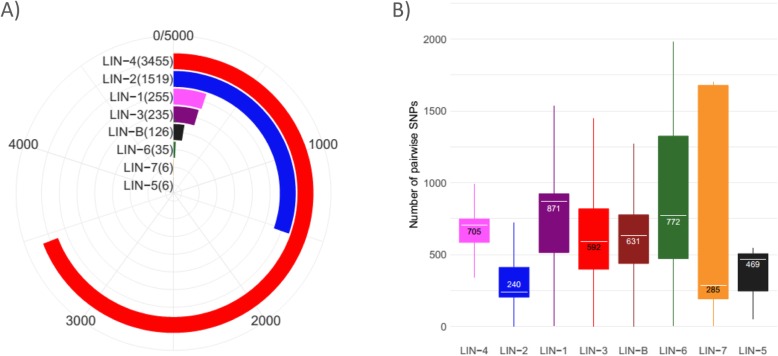
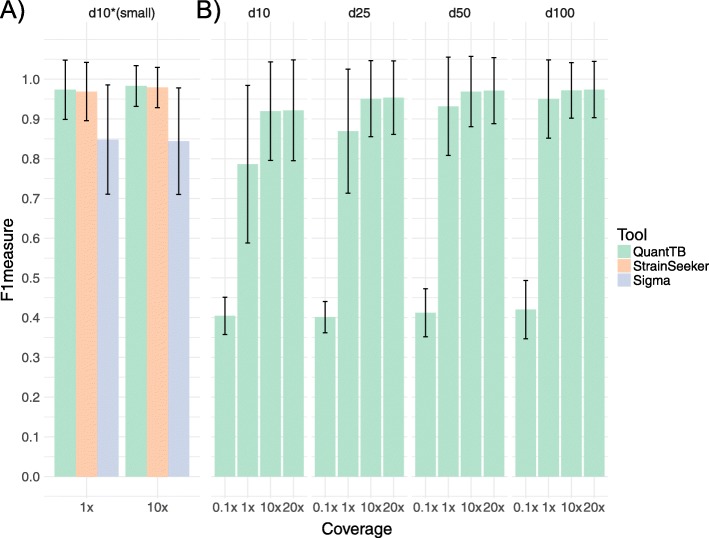
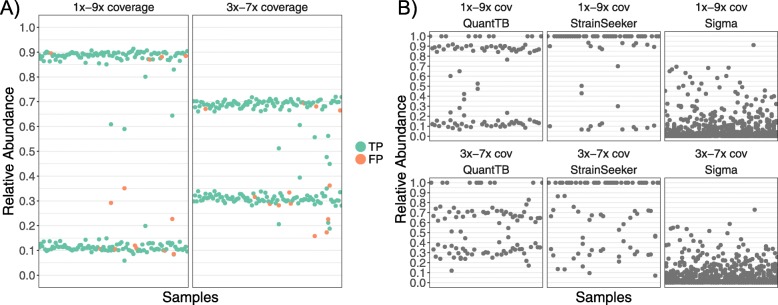
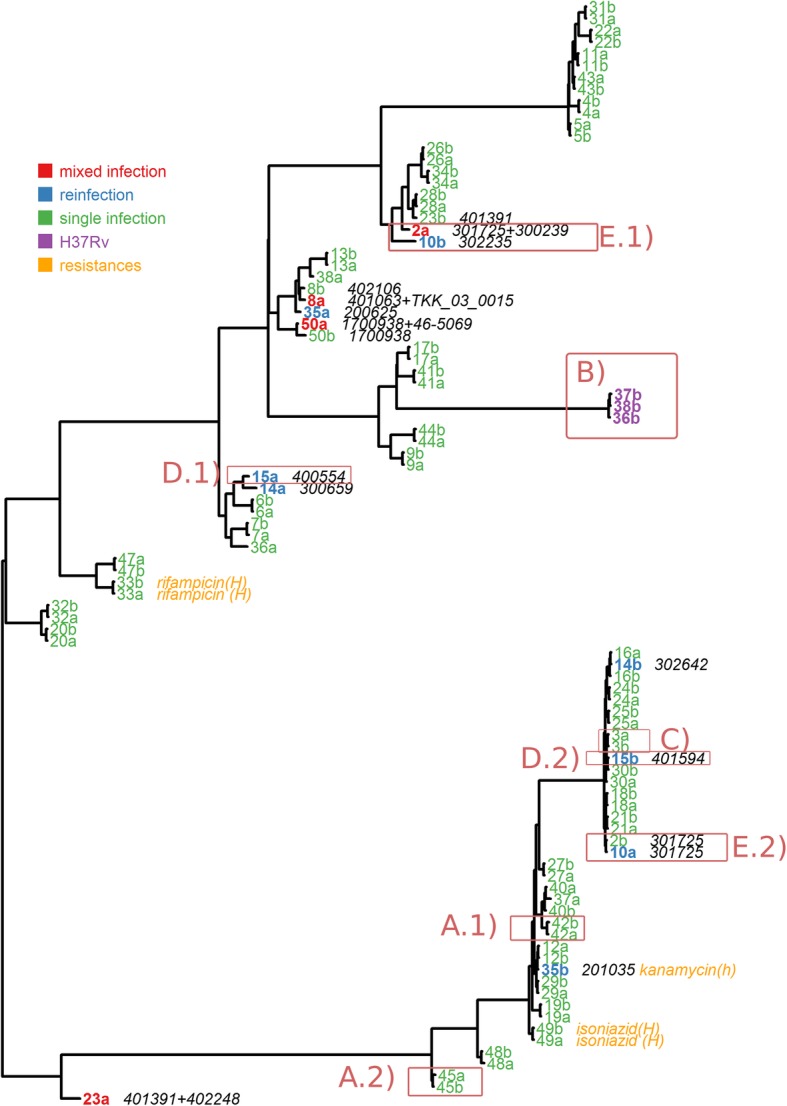
Results: We show that QuantTB has a high degree of resolution and is capable of differentiating communities differing by less than 25 SNPs and identifying strains down to 1× coverage. Using simulated data, we found QuantTB outperformed other metagenomic strain identification tools at detecting strains and quantifying strain multiplicity. In a real-world scenario, using a dataset of 50 paired clinical isolates from a study of patients with either reinfections or relapses, we found that QuantTB could detect mixed infections and reinfections at rates concordant with a manually curated approach.
Conclusion: QuantTB can determine infection multiplicity, identify hetero-resistance patterns, enable differentiation between relapse and re-infection, and clarify transmission events across seemingly unrelated patients - even in low-coverage (1×) samples. QuantTB outperforms existing tools and promises to serve as a valuable resource for both clinicians and researchers working with clinical TB samples.
Keywords: Bioinformatics; Metagenomics; Mixed infection; Mycobacterium tuberculosis; Reinfection; Strain identification; Strain level classification; Transmission; Tuberculosis; Whole genome sequencing.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- World Health Organization . Tuberculosis Fact Sheet. 2018.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
