

A CRISPR-Assisted Nonhomologous End-Joining Strategy for Efficient Genome Editing in Mycobacterium tuberculosis
- PMID: 31992616
- PMCID: PMC6989103
- DOI: 10.1128/mBio.02364-19
A CRISPR-Assisted Nonhomologous End-Joining Strategy for Efficient Genome Editing in Mycobacterium tuberculosis
Abstract
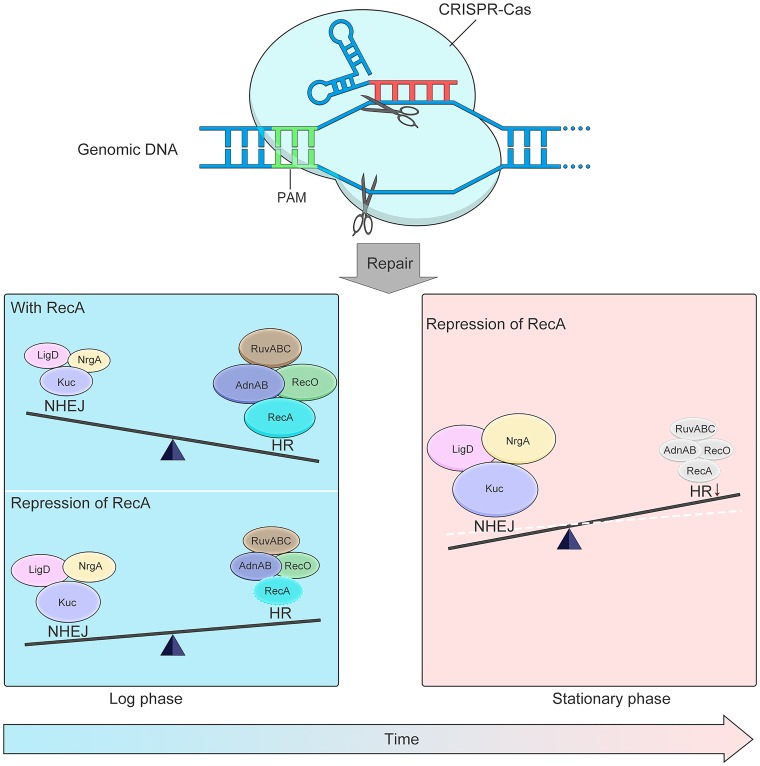
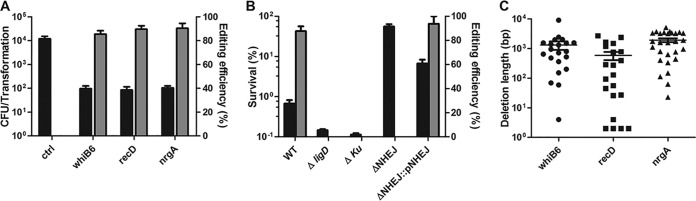
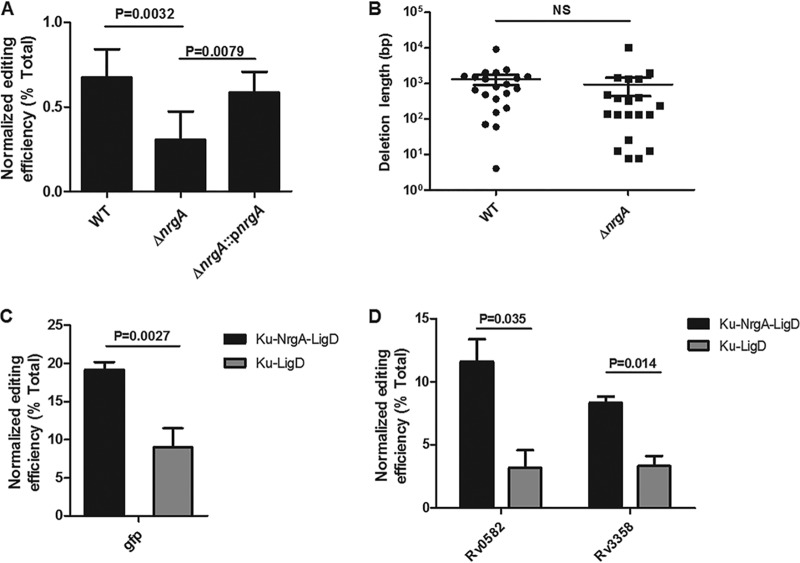
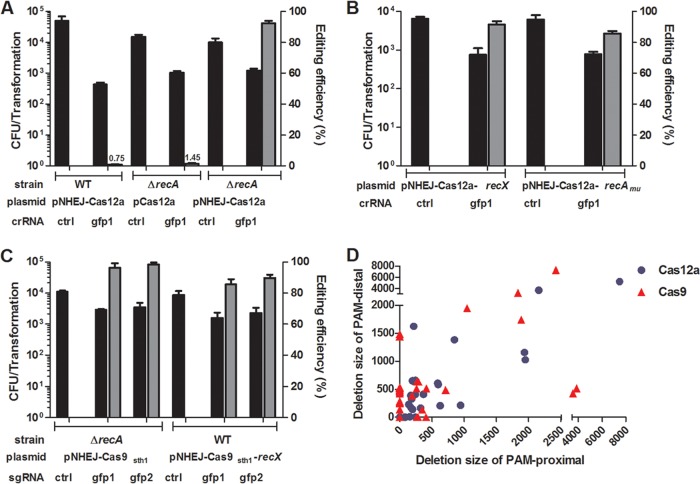
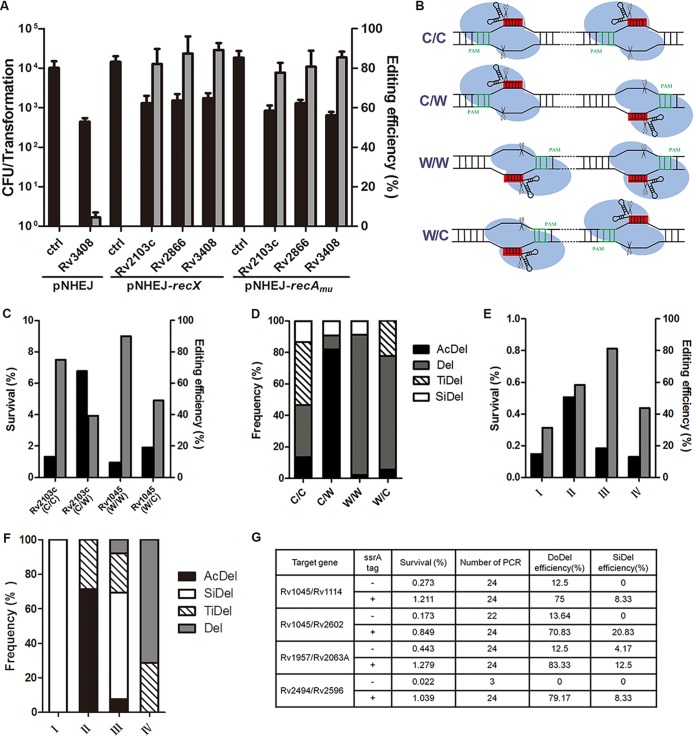
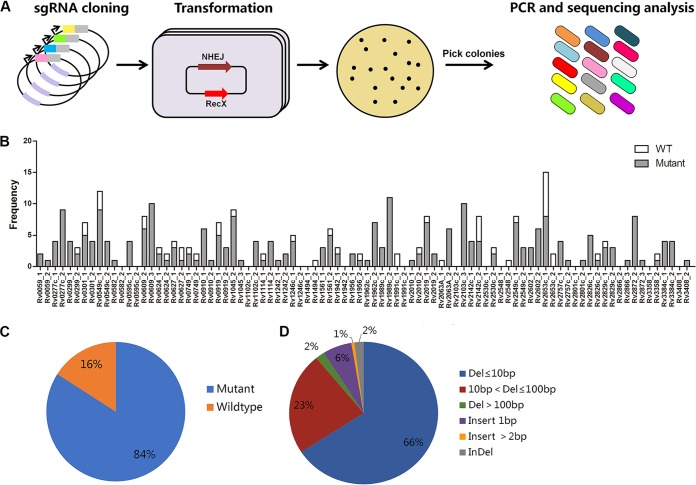
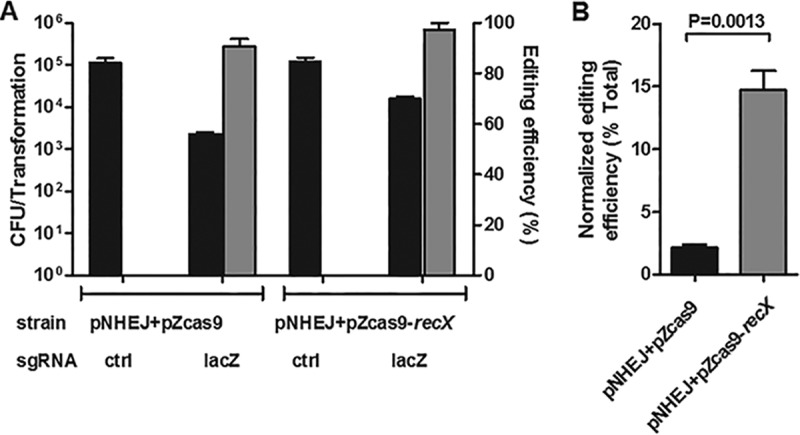
New tools for genetic manipulation of Mycobacterium tuberculosis are needed for the development of new drug regimens and vaccines aimed at curing tuberculosis infections. Clustered regularly interspaced short palindromic repeat (CRISPR)-CRISPR-associated protein (Cas) systems generate a highly specific double-strand break at the target site that can be repaired via nonhomologous end joining (NHEJ), resulting in the desired genome alteration. In this study, we first improved the NHEJ repair pathway and developed a CRISPR-Cas-mediated genome-editing method that allowed us to generate markerless deletion in Mycobacterium smegmatis, Mycobacterium marinum, and M. tuberculosis Then, we demonstrated that this system could efficiently achieve simultaneous generation of double mutations and large-scale genetic mutations in M. tuberculosis Finally, we showed that the strategy we developed can also be used to facilitate genome editing in Escherichia coli IMPORTANCE The global health impact of M. tuberculosis necessitates the development of new genetic tools for its manipulation, to facilitate the identification and characterization of novel drug targets and vaccine candidates. Clustered regularly interspaced short palindromic repeat (CRISPR)-CRISPR-associated protein (Cas) genome editing has proven to be a powerful genetic tool in various organisms; to date, however, attempts to use this approach in M. tuberculosis have failed. Here, we describe a genome-editing tool based on CRISPR cleavage and the nonhomologous end-joining (NHEJ) repair pathway that can efficiently generate deletion mutants in M. tuberculosis More importantly, this system can generate simultaneous double mutations and large-scale genetic mutations in this species. We anticipate that this CRISPR-NHEJ-assisted genome-editing system will be broadly useful for research on mycobacteria, vaccine development, and drug target profiling.
Keywords: CRISPR-Cas system; Mycobacterium marinum; Mycobacterium smegmatis; Mycobacterium tuberculosis; genome editing; nonhomologous end joining.
Copyright © 2020 Yan et al.
Figures
Similar articles
-
A CRISPR-Cpf1-Assisted Non-Homologous End Joining Genome Editing System of Mycobacterium smegmatis.Biotechnol J. 2018 Sep;13(9):e1700588. doi: 10.1002/biot.201700588. Epub 2018 Aug 6. Biotechnol J. 2018. PMID: 30039929
-
Identification of Mycobacterium tuberculosis intracellular survival-related virulence factors via CRISPR-based eukaryotic-like secretory protein mutant library screen.Microbiol Spectr. 2025 Aug 5;13(8):e0076725. doi: 10.1128/spectrum.00767-25. Epub 2025 Jun 12. Microbiol Spectr. 2025. PMID: 40503836 Free PMC article.
-
Efficient genome editing in pathogenic mycobacteria using Streptococcus thermophilus CRISPR1-Cas9.Tuberculosis (Edinb). 2020 Sep;124:101983. doi: 10.1016/j.tube.2020.101983. Epub 2020 Aug 12. Tuberculosis (Edinb). 2020. PMID: 32829077 Free PMC article.
-
Strategies for Applying Nonhomologous End Joining-Mediated Genome Editing in Prokaryotes.ACS Synth Biol. 2019 Oct 18;8(10):2194-2202. doi: 10.1021/acssynbio.9b00179. Epub 2019 Sep 26. ACS Synth Biol. 2019. PMID: 31525995 Review.
-
CRISPR-Cas systems: ushering in the new genome editing era.Bioengineered. 2018;9(1):214-221. doi: 10.1080/21655979.2018.1470720. Bioengineered. 2018. PMID: 29968520 Free PMC article. Review.
Cited by
-
A CRISPR-guided mutagenic DNA polymerase strategy for the detection of antibiotic-resistant mutations in M. tuberculosis.Mol Ther Nucleic Acids. 2022 Jul 12;29:354-367. doi: 10.1016/j.omtn.2022.07.004. eCollection 2022 Sep 13. Mol Ther Nucleic Acids. 2022. PMID: 35950213 Free PMC article.
-
Modifying Escherichia coli to mimic Shigella for medical microbiology laboratory teaching: a new strategy to improve biosafety in class.Front Cell Infect Microbiol. 2023 Sep 13;13:1257361. doi: 10.3389/fcimb.2023.1257361. eCollection 2023. Front Cell Infect Microbiol. 2023. PMID: 37780843 Free PMC article.
-
Genetic tools for the development of recombinant lactic acid bacteria.Microb Cell Fact. 2021 Jun 19;20(1):118. doi: 10.1186/s12934-021-01607-1. Microb Cell Fact. 2021. PMID: 34147119 Free PMC article. Review.
-
Challenges in delivery systems for CRISPR-based genome editing and opportunities of nanomedicine.Biomed Eng Lett. 2021 Jul 13;11(3):217-233. doi: 10.1007/s13534-021-00199-4. eCollection 2021 Aug. Biomed Eng Lett. 2021. PMID: 34350049 Free PMC article. Review.
-
CRISPR Interference (CRISPRi) for Targeted Gene Silencing in Mycobacteria.Methods Mol Biol. 2021;2314:343-364. doi: 10.1007/978-1-0716-1460-0_16. Methods Mol Biol. 2021. PMID: 34235662
References
-
- World Health Organization. 2018. Global tuberculosis report 2018. WHO, Geneva, Switzerland.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
