

Genome-wide rare variant analysis for thousands of phenotypes in over 70,000 exomes from two cohorts
- PMID: 31992710
- PMCID: PMC6987107
- DOI: 10.1038/s41467-020-14288-y
Genome-wide rare variant analysis for thousands of phenotypes in over 70,000 exomes from two cohorts
Abstract
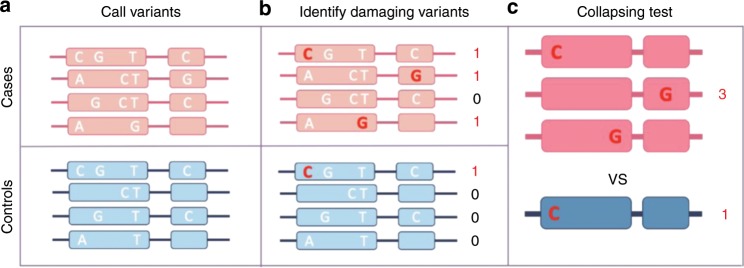
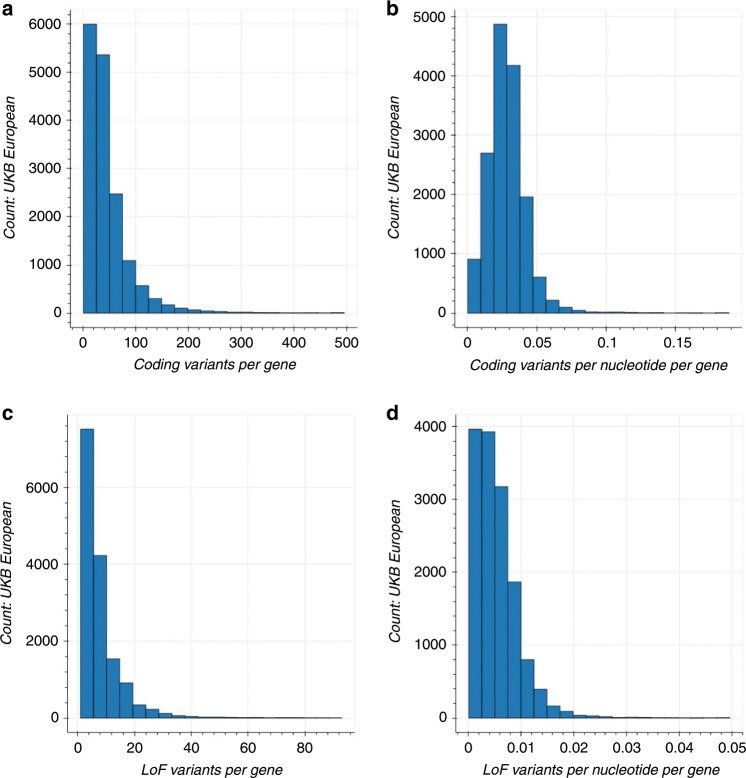
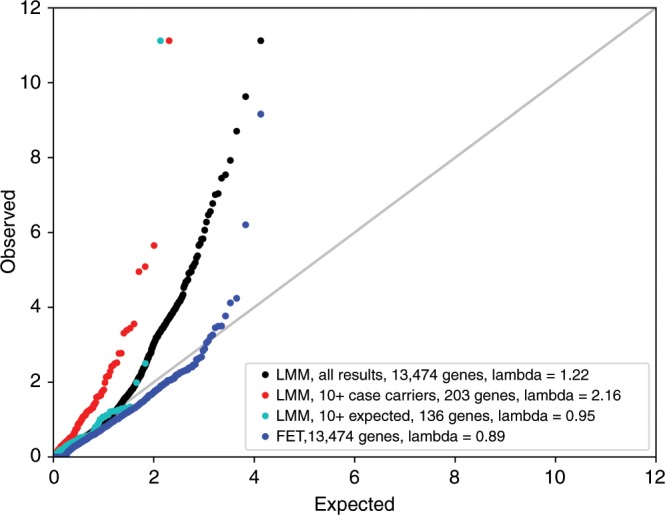
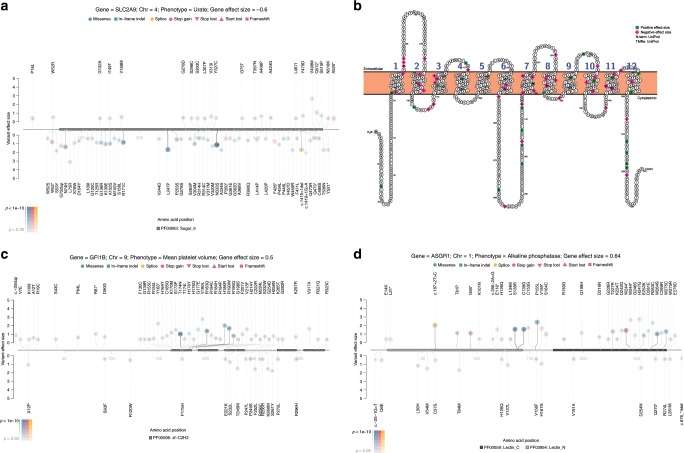
Understanding the impact of rare variants is essential to understanding human health. We analyze rare (MAF < 0.1%) variants against 4264 phenotypes in 49,960 exome-sequenced individuals from the UK Biobank and 1934 phenotypes (1821 overlapping with UK Biobank) in 21,866 members of the Healthy Nevada Project (HNP) cohort who underwent Exome + sequencing at Helix. After using our rare-variant-tailored methodology to reduce test statistic inflation, we identify 64 statistically significant gene-based associations in our meta-analysis of the two cohorts and 37 for phenotypes available in only one cohort. Singletons make significant contributions to our results, and the vast majority of the associations could not have been identified with a genotyping chip. Our results are available for interactive browsing in a webapp (https://ukb.research.helix.com). This comprehensive analysis illustrates the biological value of large, deeply phenotyped cohorts of unselected populations coupled with NGS data.
Conflict of interest statement
E.T.C., S.W., N.L.W., F.T., D.M.F., E.S., M.I. and J.T.L. are employees of Helix. R.W.R., G.E., W.J.M., K.A.S. and J.J.G. declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
