

A mouse model for kinesin family member 11 (Kif11)-associated familial exudative vitreoretinopathy
- PMID: 31993640
- PMCID: PMC7206855
- DOI: 10.1093/hmg/ddaa018
A mouse model for kinesin family member 11 (Kif11)-associated familial exudative vitreoretinopathy
Abstract
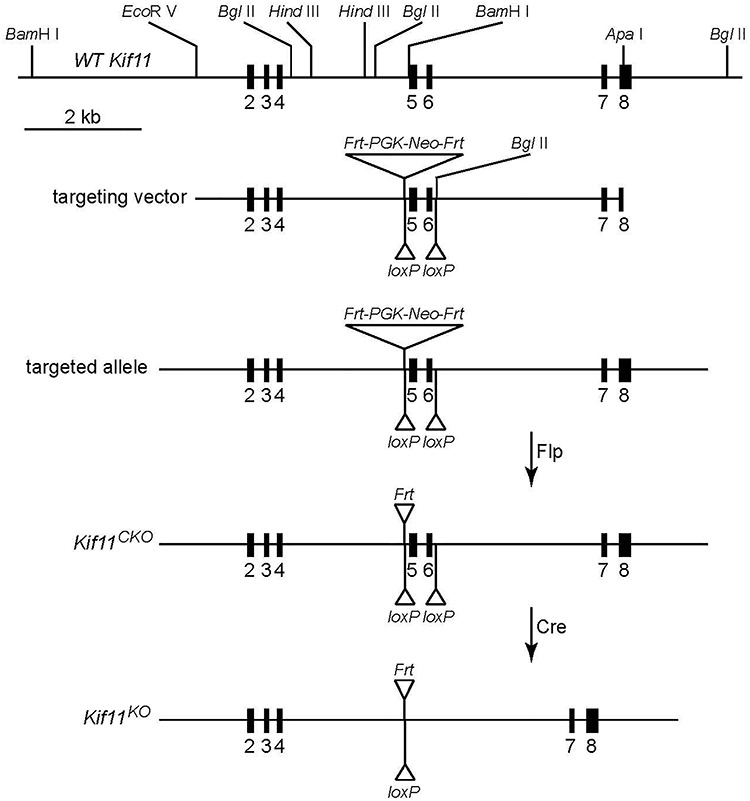
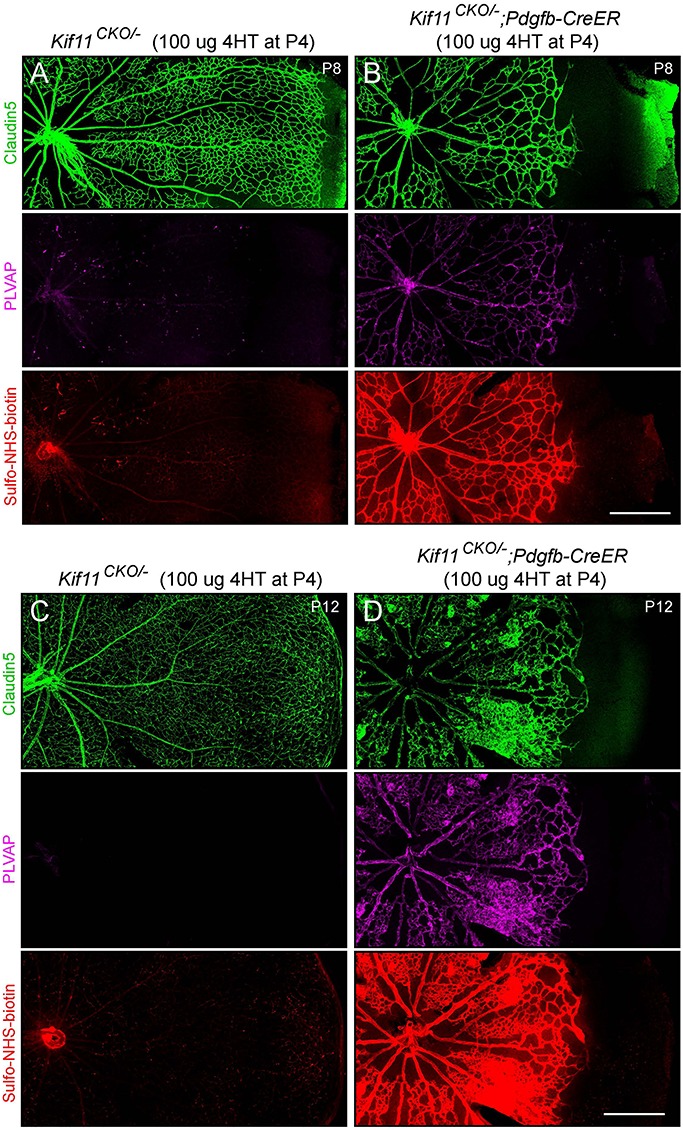
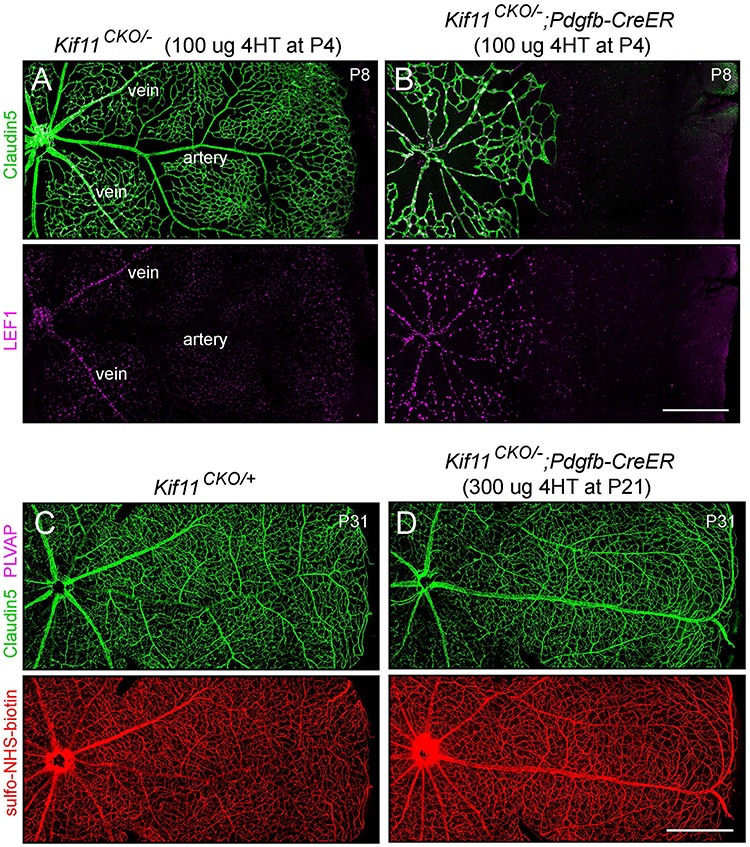
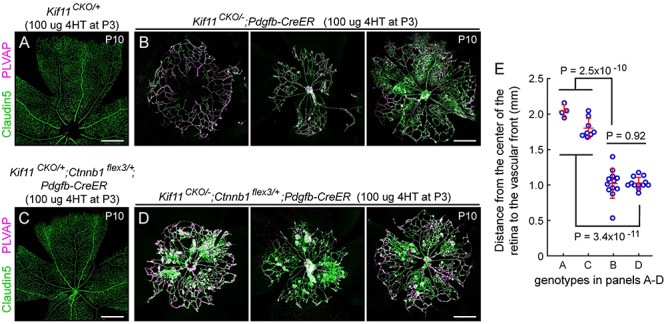
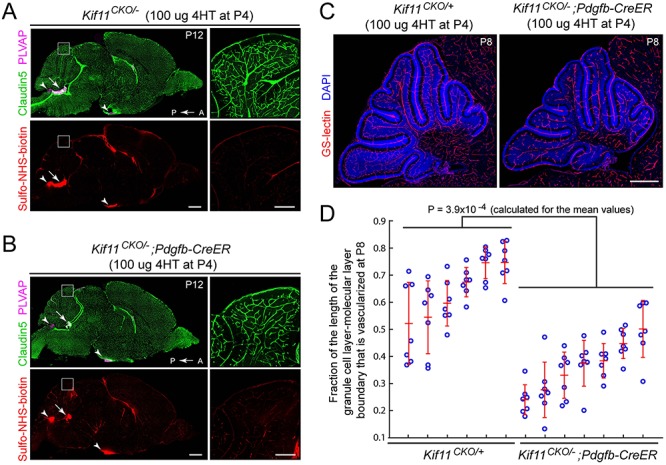
During mitosis, Kif11, a kinesin motor protein, promotes bipolar spindle formation and chromosome movement, and during interphase, Kif11 mediates diverse trafficking processes in the cytoplasm. In humans, inactivating mutations in KIF11 are associated with (1) retinal hypovascularization with or without microcephaly and (2) multi-organ syndromes characterized by variable combinations of lymphedema, chorioretinal dysplasia, microcephaly and/or mental retardation. To explore the pathogenic basis of KIF11-associated retinal vascular disease, we generated a Kif11 conditional knockout (CKO) mouse and investigated the consequences of early postnatal inactivation of Kif11 in vascular endothelial cells (ECs). The principal finding is that postnatal EC-specific loss of Kif11 leads to severely stunted growth of the retinal vasculature, mildly stunted growth of the cerebellar vasculature and little or no effect on the vasculature elsewhere in the central nervous system (CNS). Thus, in mice, Kif11 function in early postnatal CNS ECs is most significant in the two CNS regions-the retina and cerebellum-that exhibit the most rapid rate of postnatal growth, which may sensitize ECs to impaired mitotic spindle function. Several lines of evidence indicate that these phenotypes are not caused by reduced beta-catenin signaling in ECs, despite the close resemblance of the Kif11 CKO phenotype to that caused by EC-specific reductions in beta-catenin signaling. Based on prior work, defective beta-catenin signaling had been the only known mechanism responsible for monogenic human disorders of retinal hypovascularization. The present study implies that retinal hypovascularization can arise from a second and mechanistically distinct cause.
© The Author(s) 2020. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Nikopoulos K., Venselaar H., Collin R.W., Riveiro-Alvarez R., Boonstra F.N., Hooymans J.M., Mukhopadhyay A., Shears D., van Bers M., de Wijs I.J. et al. (2010) Overview of the mutation spectrum in familial exudative vitreoretinopathy and Norrie disease with identification of 21 novel variants in FZD4, LRP5, and NDP. Hum. Mutat., 31, 656–666. - PubMed
-
- Rao F.Q., Cai X.B., Cheng F.F., Cheng W., Fang X.L., Li N., Huang X.F., Li L.H. and Jin Z.B. (2017) Mutations in LRP5,FZD4, TSPAN12, NDP, ZNF408, or KIF11 genes account for 38.7% of Chinese patients with familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci., 58, 2623–2629. - PubMed
-
- Tang M., Sun L., Hu A., Yuan M., Yang Y., Peng X. and Ding X. (2017) Mutation spectrum of the LRP5, NDP, and TSPAN12 genes in Chinese patients with familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci., 58, 5949–5957. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
