

Remethylation of Dnmt3a-/- hematopoietic cells is associated with partial correction of gene dysregulation and reduced myeloid skewing
- PMID: 31996479
- PMCID: PMC7022185
- DOI: 10.1073/pnas.1918611117
Remethylation of Dnmt3a-/- hematopoietic cells is associated with partial correction of gene dysregulation and reduced myeloid skewing
Abstract
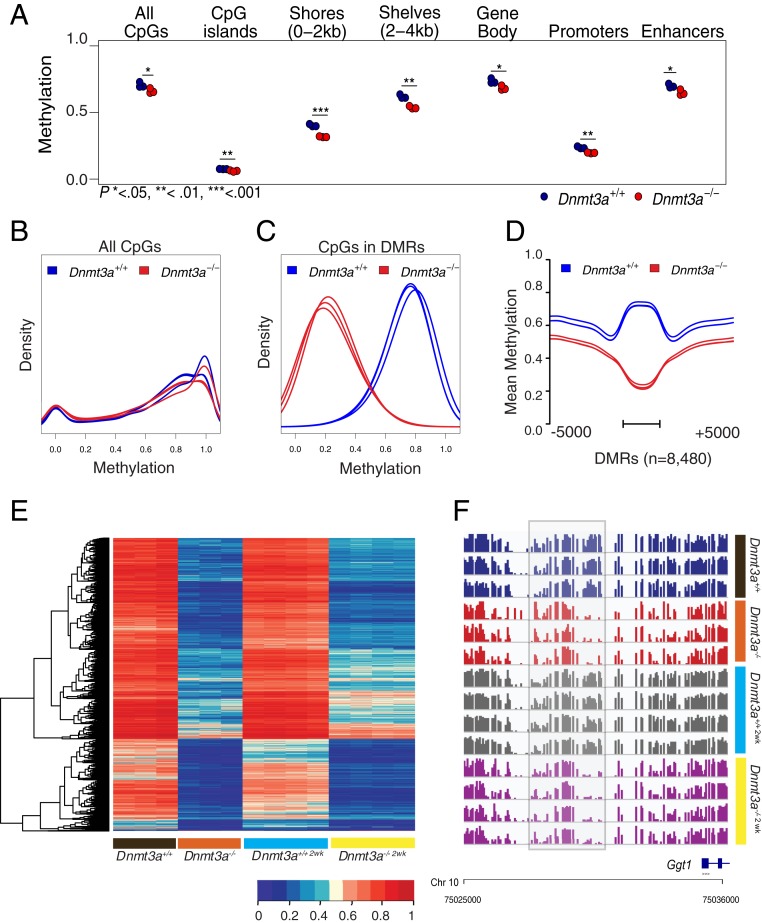
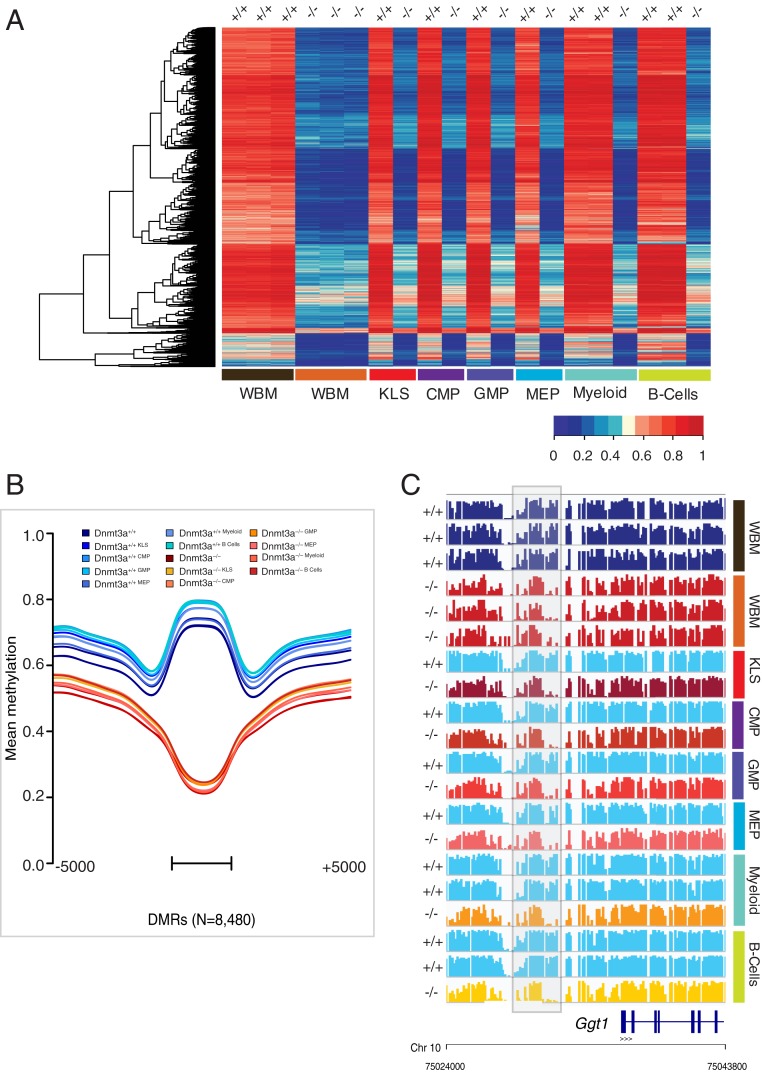
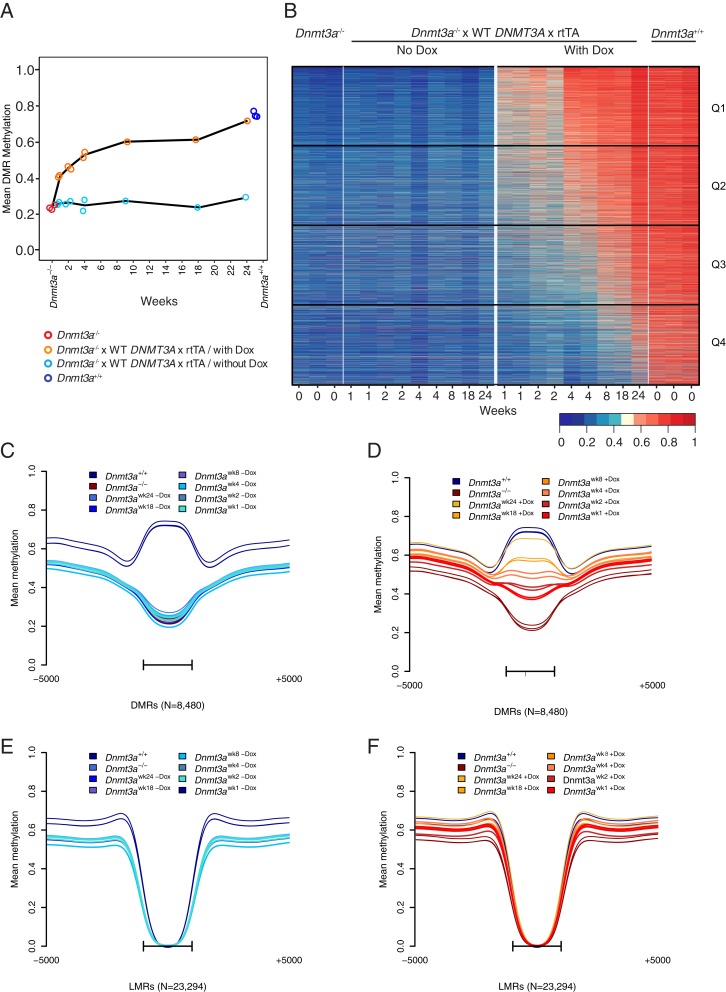
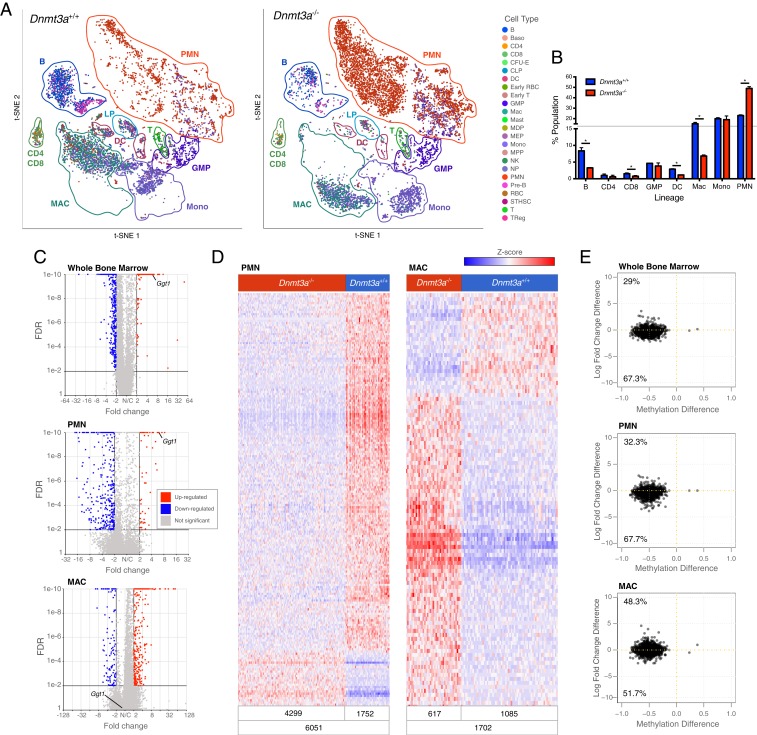
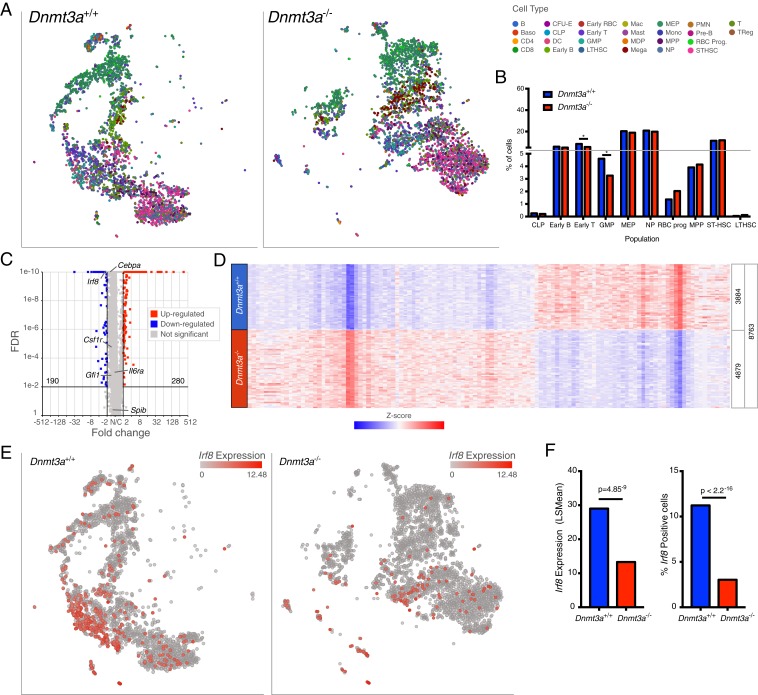
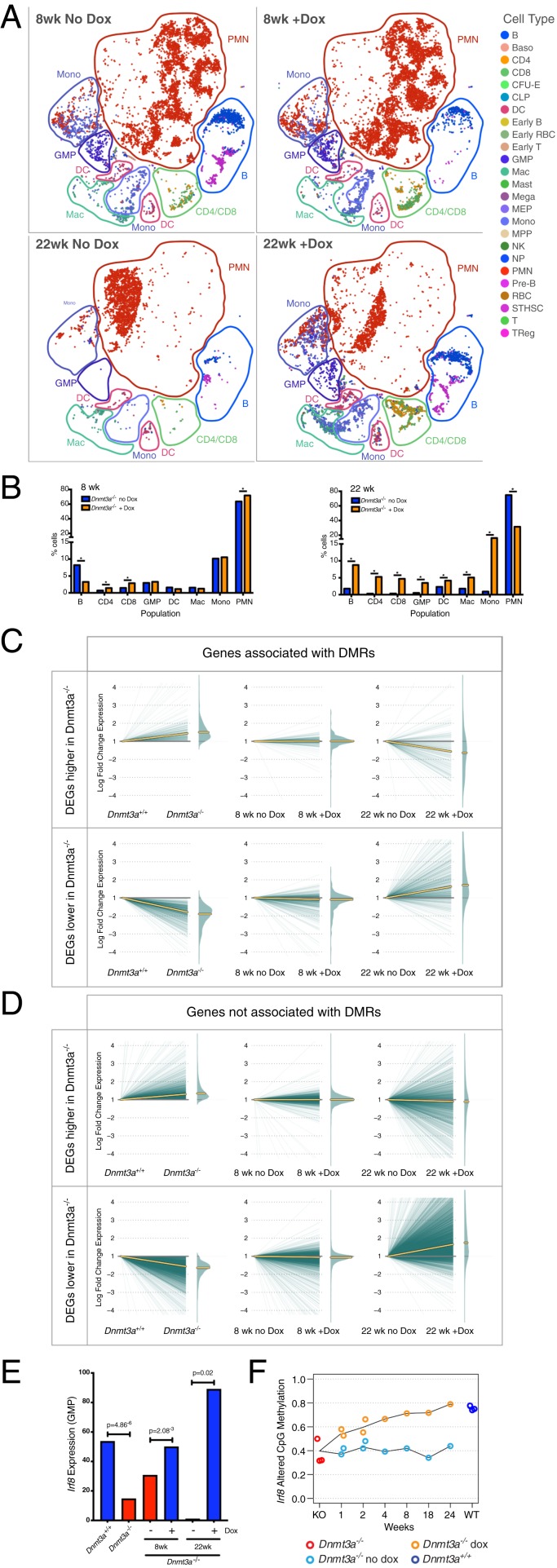
Mutations in the DNA methyltransferase 3A (DNMT3A) gene are the most common cause of age-related clonal hematopoiesis (ARCH) in older individuals, and are among the most common initiating events for acute myeloid leukemia (AML). The most frequent DNMT3A mutation in AML patients (R882H) encodes a dominant-negative protein that reduces methyltransferase activity by ∼80% in cells with heterozygous mutations, causing a focal, canonical DNA hypomethylation phenotype; this phenotype is partially recapitulated in murine Dnmt3a-/- bone marrow cells. To determine whether the hypomethylation phenotype of Dnmt3a-/- hematopoietic cells is reversible, we developed an inducible transgene to restore expression of DNMT3A in transplanted bone marrow cells from Dnmt3a-/- mice. Partial remethylation was detected within 1 wk, but near-complete remethylation required 6 mo. Remethylation was accurate, dynamic, and highly ordered, suggesting that differentially methylated regions have unique properties that may be relevant for their functions. Importantly, 22 wk of DNMT3A addback partially corrected dysregulated gene expression, and mitigated the expansion of myeloid cells. These data show that restoring DNMT3A expression can alter the epigenetic "state" created by loss of Dnmt3a activity; this genetic proof-of-concept experiment suggests that this approach could be relevant for patients with ARCH or AML caused by loss-of-function DNMT3A mutations.
Keywords: DNA methylation; DNA methyltransferase; gene expression; hematopoiesis.
Copyright © 2020 the Author(s). Published by PNAS.
Conflict of interest statement
Competing interest statement: T.J.L. is engaged in a scientific collaboration with Rigel Pharmaceuticals to develop drugs that inhibit the function of the DNMT3A R882H mutation. He has received two honoraria from Rigel Pharmaceuticals to give presentations at their company in South San Francisco, CA. None of this work is described in this paper.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
