

Tobacco smoking and somatic mutations in human bronchial epithelium
- PMID: 31996850
- PMCID: PMC7021511
- DOI: 10.1038/s41586-020-1961-1
Tobacco smoking and somatic mutations in human bronchial epithelium
Abstract
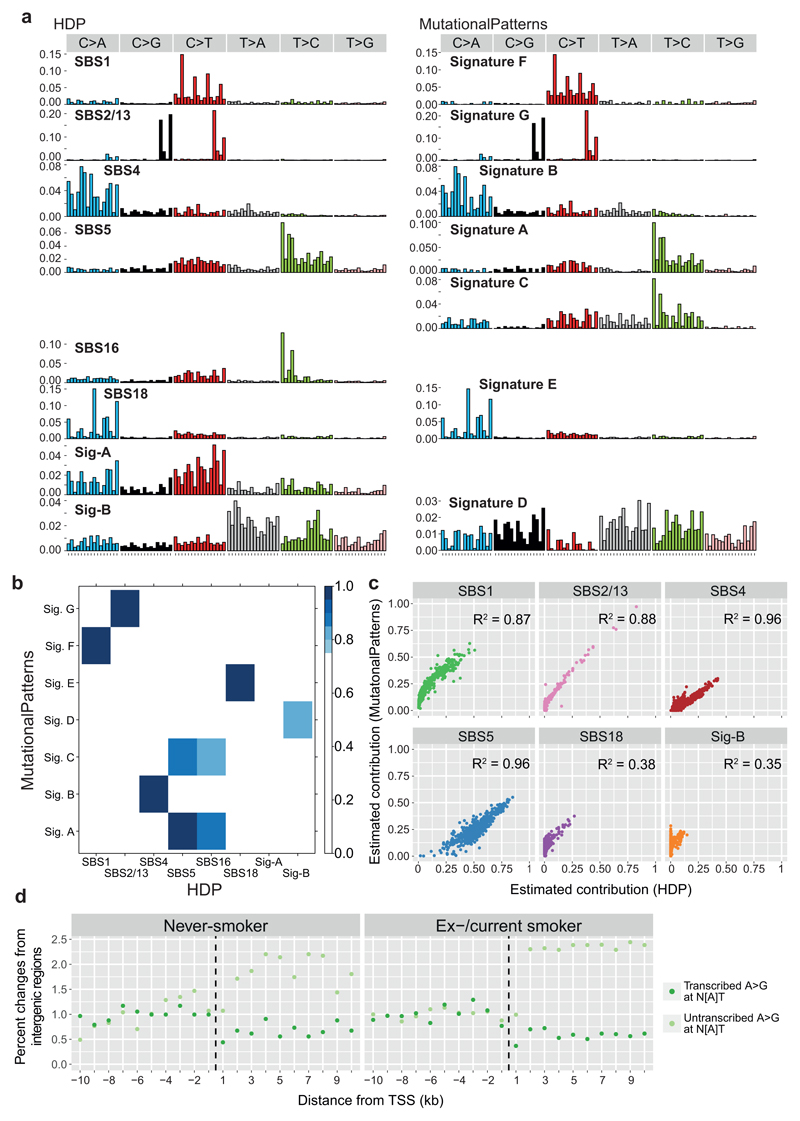
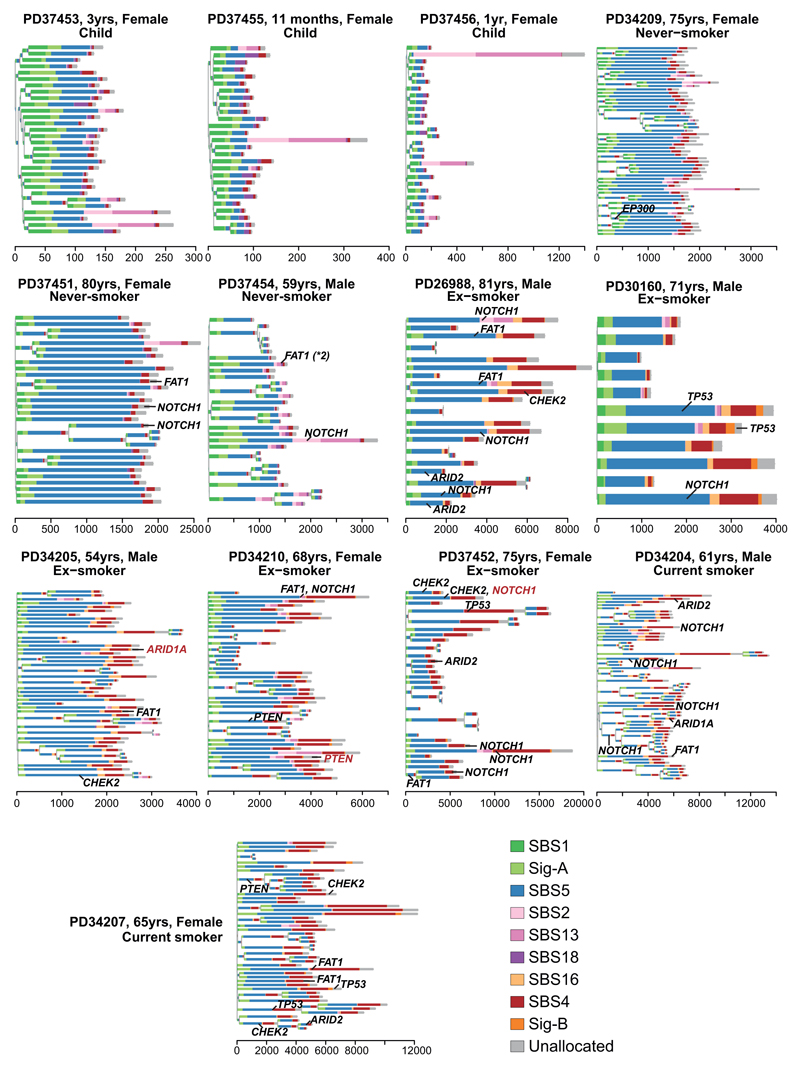
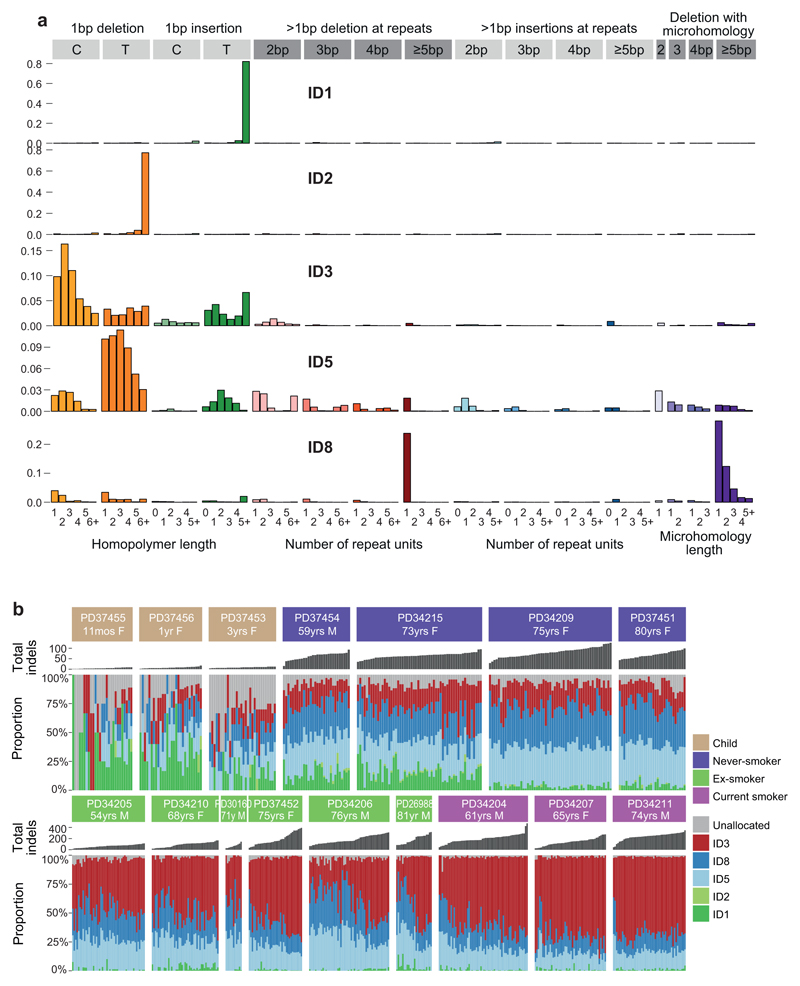
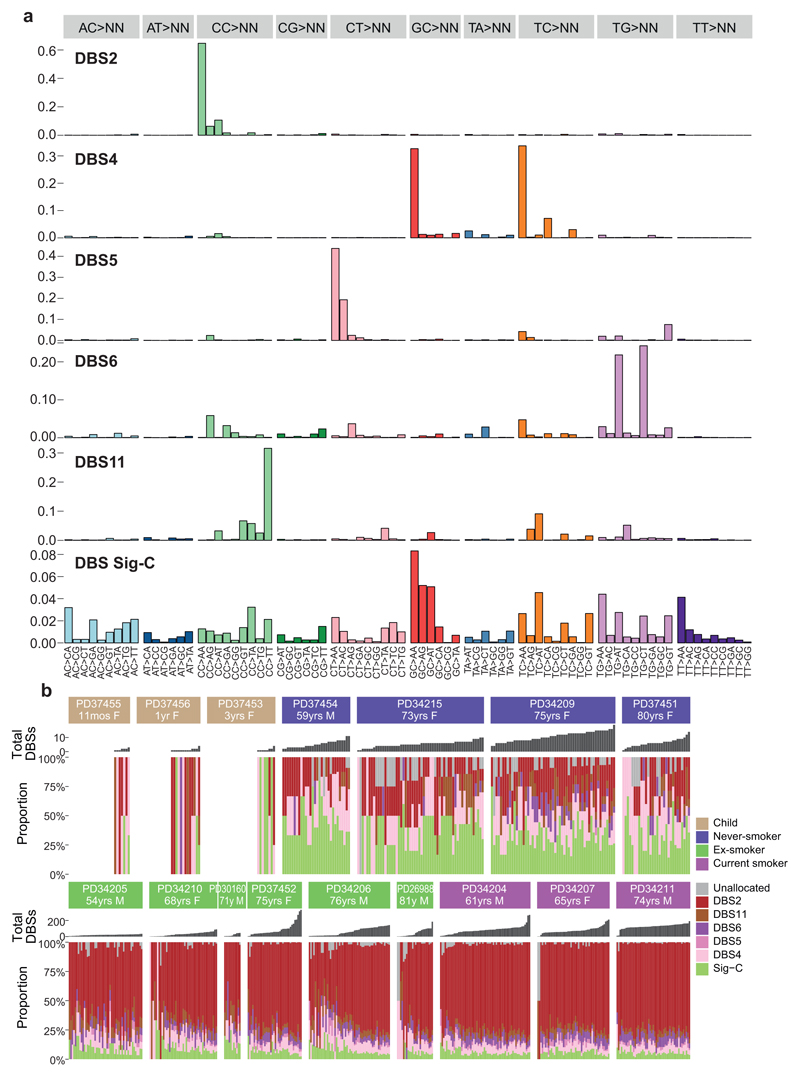
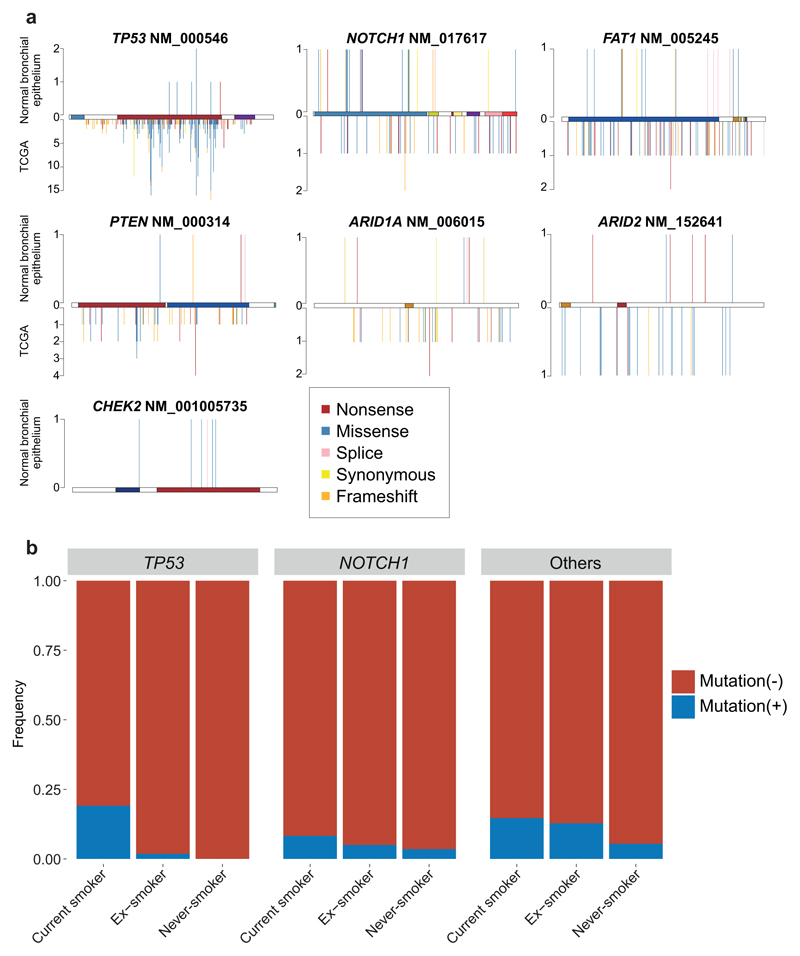
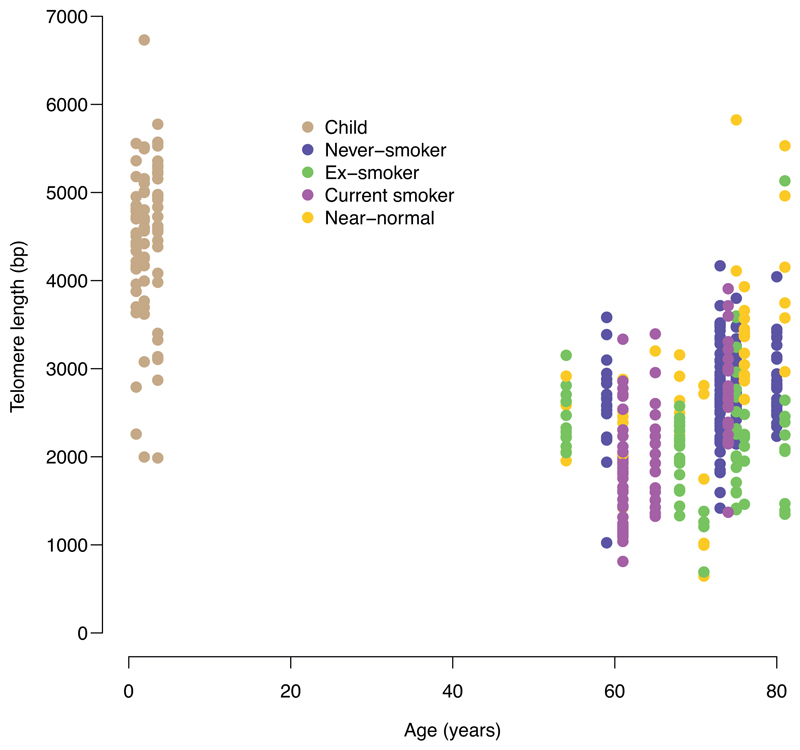
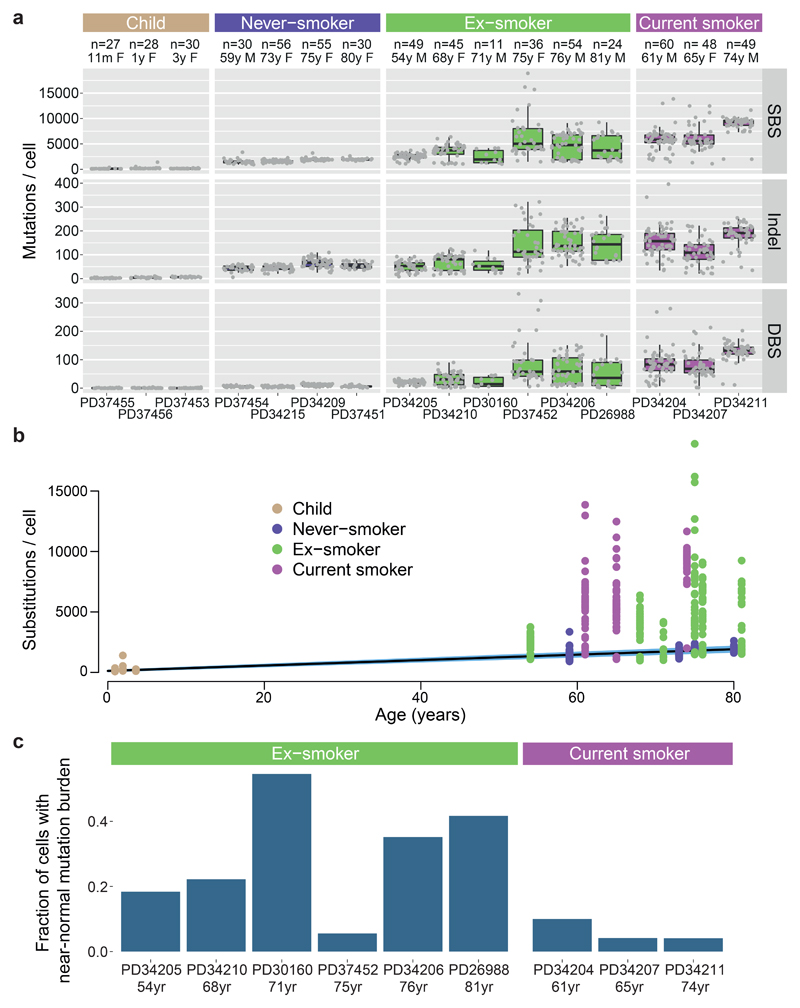
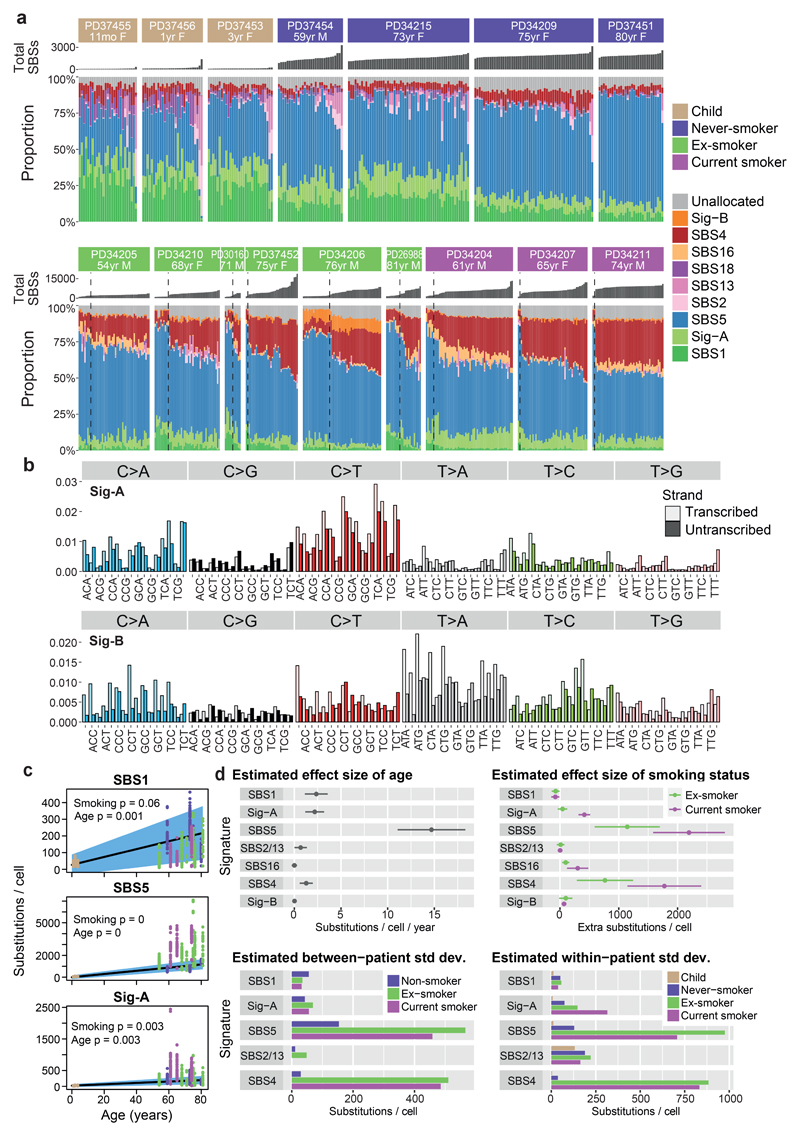
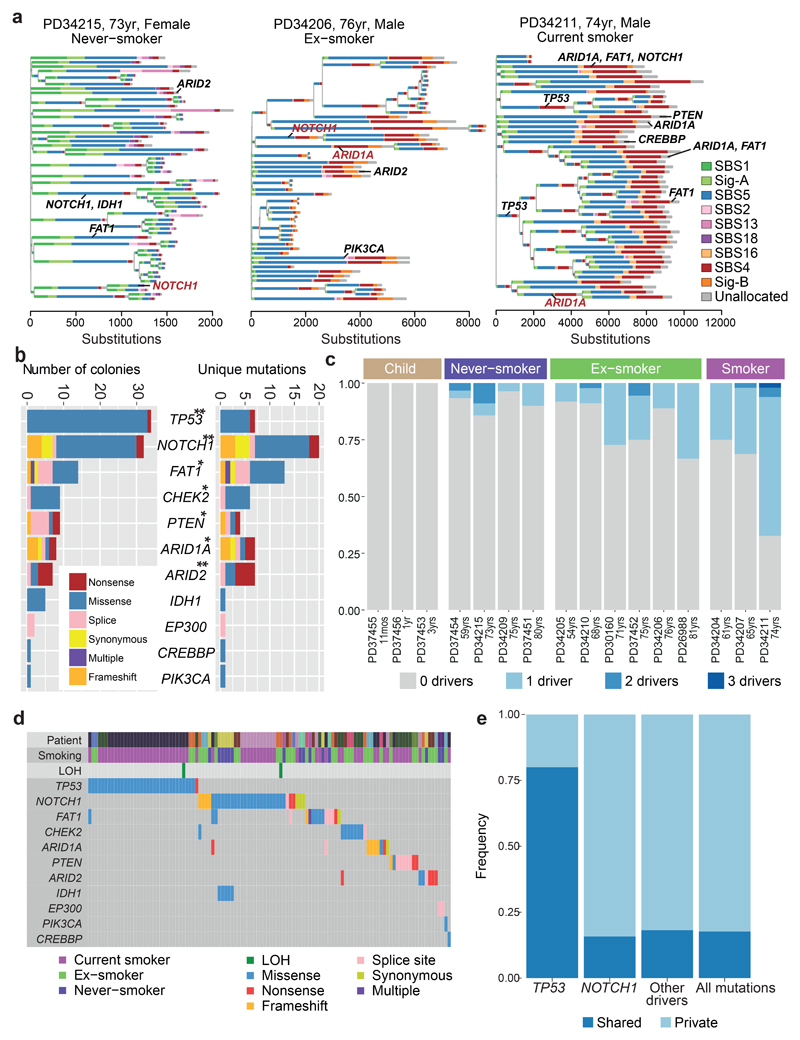
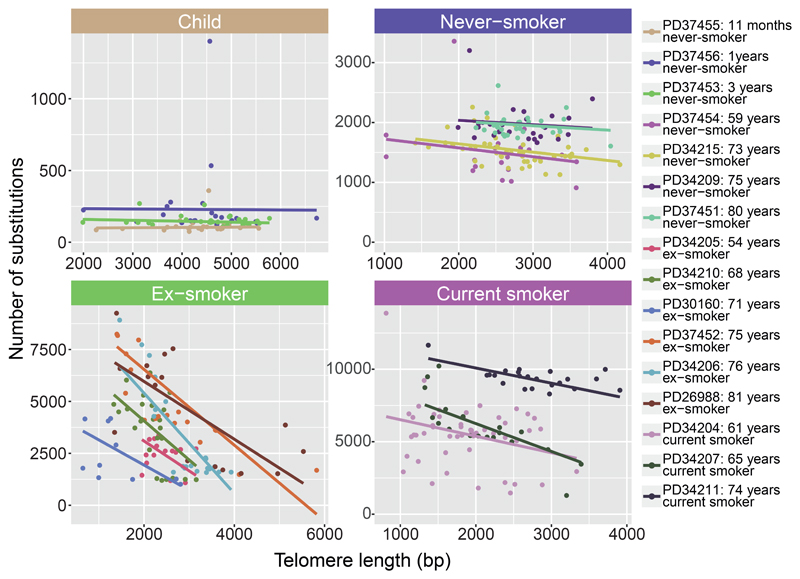
Tobacco smoking causes lung cancer1-3, a process that is driven by more than 60 carcinogens in cigarette smoke that directly damage and mutate DNA4,5. The profound effects of tobacco on the genome of lung cancer cells are well-documented6-10, but equivalent data for normal bronchial cells are lacking. Here we sequenced whole genomes of 632 colonies derived from single bronchial epithelial cells across 16 subjects. Tobacco smoking was the major influence on mutational burden, typically adding from 1,000 to 10,000 mutations per cell; massively increasing the variance both within and between subjects; and generating several distinct mutational signatures of substitutions and of insertions and deletions. A population of cells in individuals with a history of smoking had mutational burdens that were equivalent to those expected for people who had never smoked: these cells had less damage from tobacco-specific mutational processes, were fourfold more frequent in ex-smokers than current smokers and had considerably longer telomeres than their more-mutated counterparts. Driver mutations increased in frequency with age, affecting 4-14% of cells in middle-aged subjects who had never smoked. In current smokers, at least 25% of cells carried driver mutations and 0-6% of cells had two or even three drivers. Thus, tobacco smoking increases mutational burden, cell-to-cell heterogeneity and driver mutations, but quitting promotes replenishment of the bronchial epithelium from mitotically quiescent cells that have avoided tobacco mutagenesis.
Conflict of interest statement
The authors declare no competing interests.
Figures
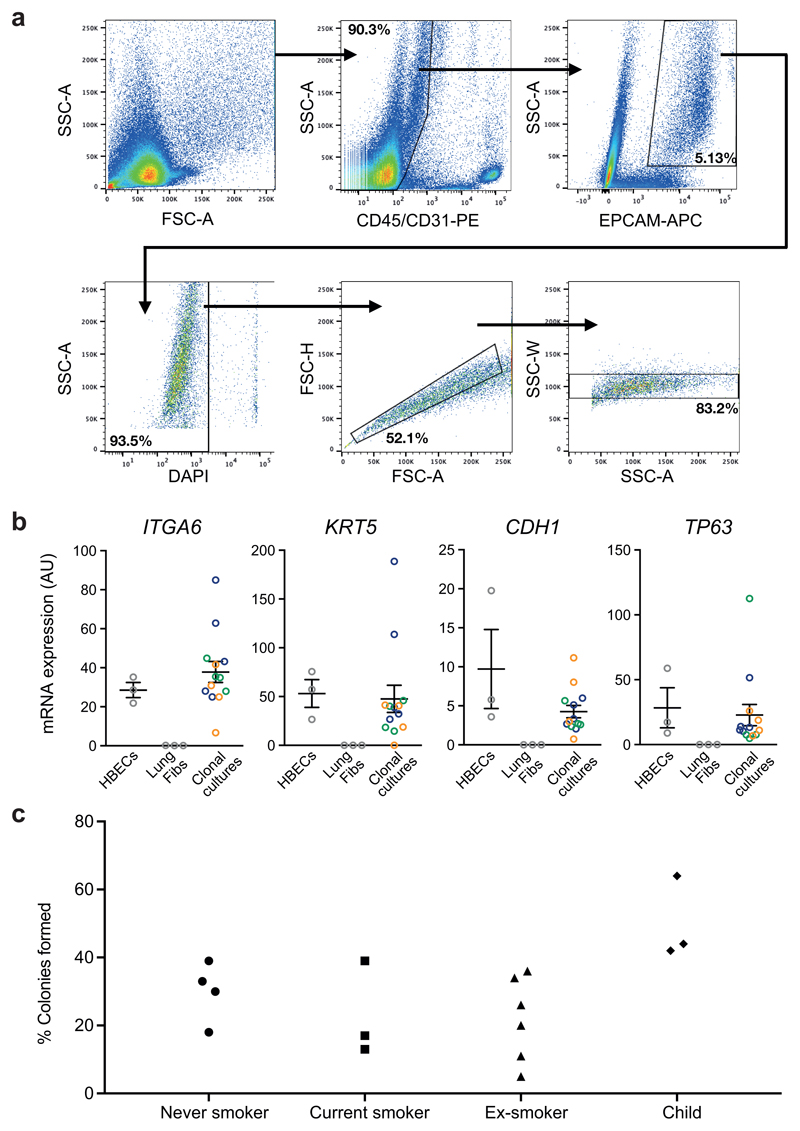
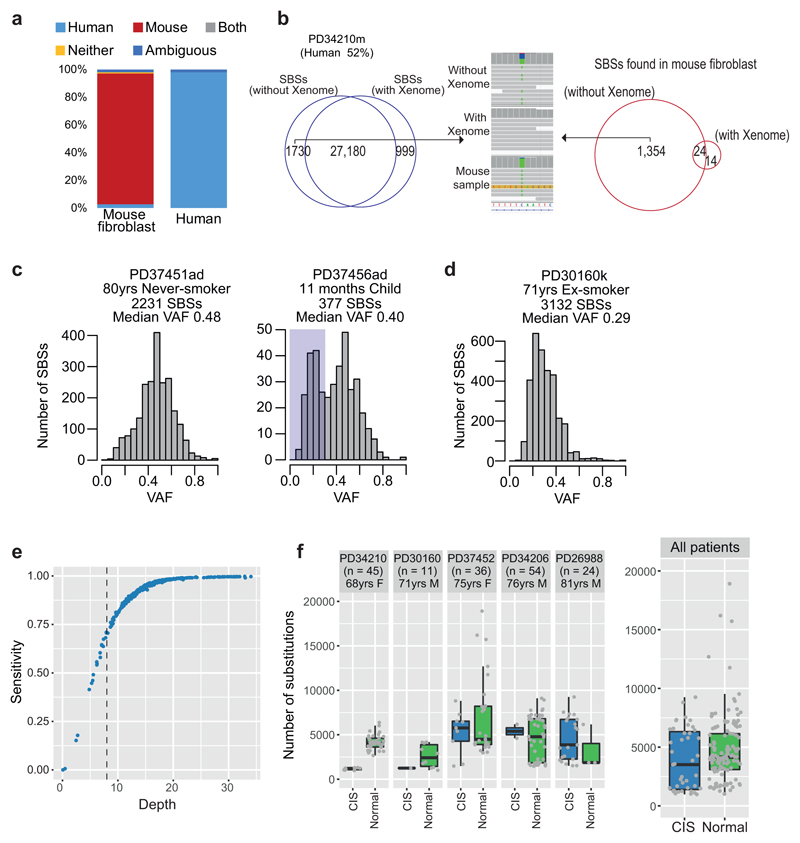
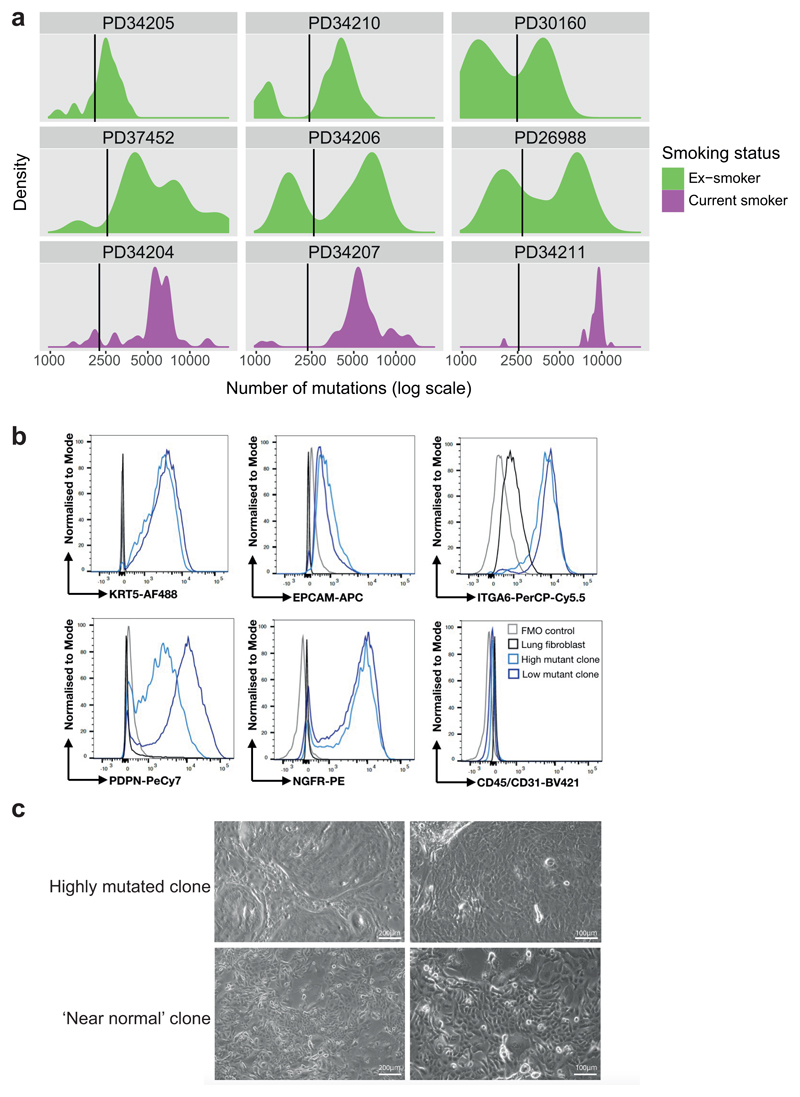
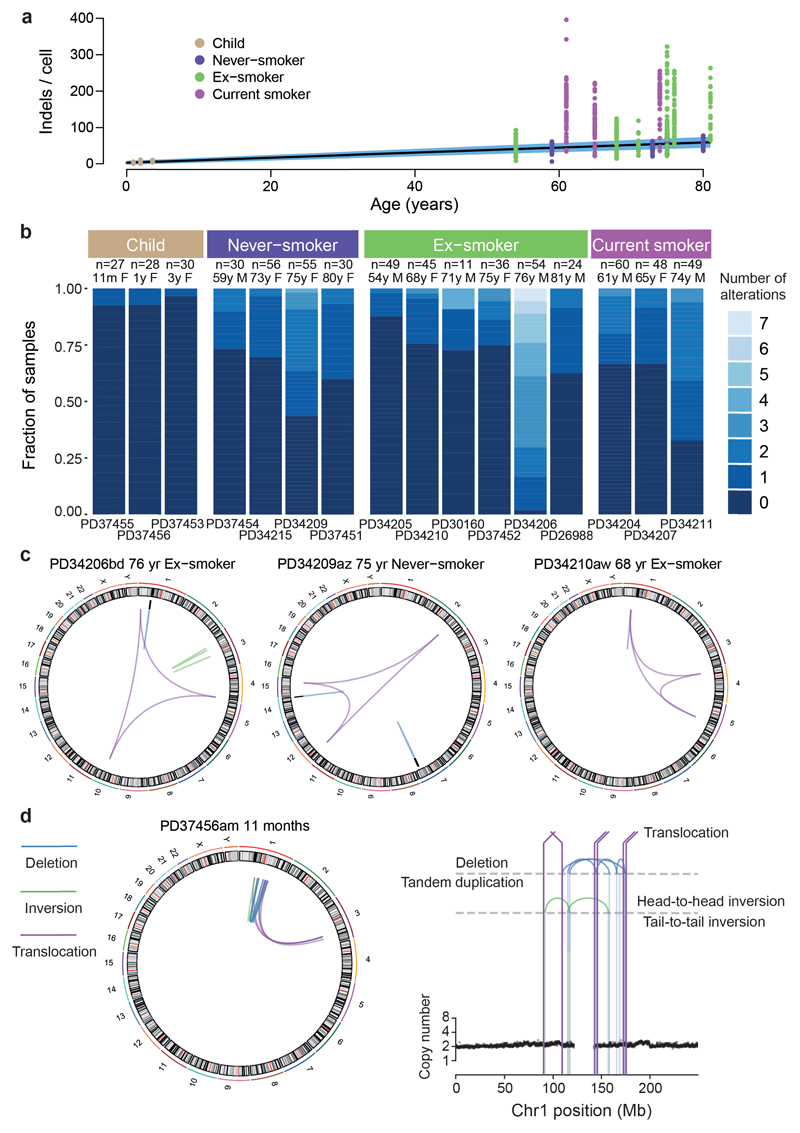
Comment in
-
Smoke signals in the DNA of normal lung cells.Nature. 2020 Feb;578(7794):224-226. doi: 10.1038/d41586-020-00165-7. Nature. 2020. PMID: 32042085 No abstract available.
-
Complex effects of tobacco on lung tissue.Nat Rev Cancer. 2020 Apr;20(4):199. doi: 10.1038/s41568-020-0249-y. Nat Rev Cancer. 2020. PMID: 32112043 No abstract available.
References
-
- International Agency for Research on Cancer. Tobacco Smoke and Involuntary Smoking. 2004;83
-
- Pfeifer GP, et al. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21:7435–7451. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
