

Improved translation of stability for conjugated antibodies using an in vitro whole blood assay
- PMID: 31997712
- PMCID: PMC6999835
- DOI: 10.1080/19420862.2020.1715705
Improved translation of stability for conjugated antibodies using an in vitro whole blood assay
Abstract
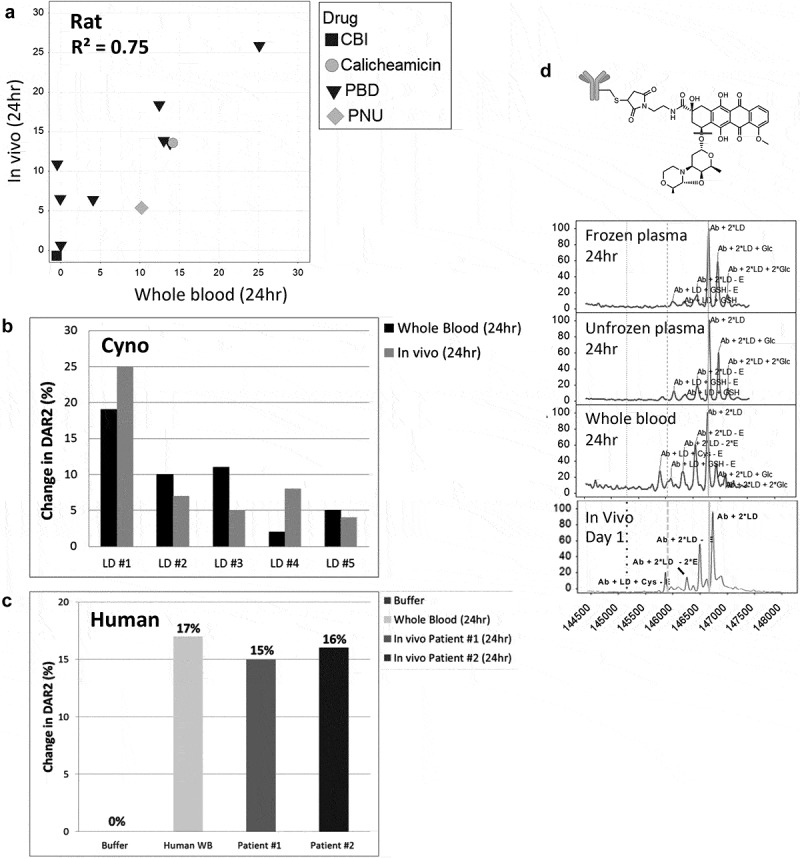
For antibody-drug conjugates to be efficacious and safe, they must be stable in circulation to carry the payload to the site of the targeted cell. Several components of a drug-conjugated antibody are known to influence stability: 1) the site of drug attachment on the antibody, 2) the linker used to attach the payload to the antibody, and 3) the payload itself. In order to support the design and optimization of a high volume of drug conjugates and avoid unstable conjugates prior to testing in animal models, we wanted to proactively identify these potential liabilities. Therefore, we sought to establish an in vitro screening method that best correlated with in vivo stability. While traditionally plasma has been used to assess in vitro stability, our evaluation using a variety of THIOMABTM antibody-drug conjugates revealed several disconnects between the stability assessed in vitro and the in vivo outcomes when using plasma. When drug conjugates were incubated in vitro for 24 h in mouse whole blood rather than plasma and then analyzed by affinity capture LC-MS, we found an improved correlation to in vivo stability with whole blood (R2 = 0.87, coefficient of determination) compared to unfrozen or frozen mouse plasma (R2 = 0.34, 0.01, respectively). We further showed that this whole blood assay was also able to predict in vivo stability of other preclinical species such as rat and cynomolgus monkey, as well as in human. The screening method utilized short (24 h) incubation times, as well as a custom analysis software, allowing increased throughput and in-depth biotransformation characterization. While some instabilities that were more challenging to identify remain, the method greatly enhanced the process of screening, optimizing, and lead candidate selection, resulting in the substantial reduction of animal studies.
Keywords: Stability; antibody-drug conjugate; drug modification; plasma; whole blood.
Figures




References
-
- Wang H, Rangan VS, Sung MC, Passmore D, Kempe T, Wang X, Thevanayagam L, Pan C, Rao C, Srinivasan M, et al. Pharmacokinetic characterization of BMS-936561, an anti-CD70 antibody-drug conjugate, in preclinical animal species and prediction of its pharmacokinetics in humans. Biopharm Drug Dispos. 2016. March;37(2):93–106. doi:10.1002/bdd.1953. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources