

Potential TMA-Producing Bacteria Are Ubiquitously Found in Mammalia
- PMID: 31998260
- PMCID: PMC6964529
- DOI: 10.3389/fmicb.2019.02966
Potential TMA-Producing Bacteria Are Ubiquitously Found in Mammalia
Abstract
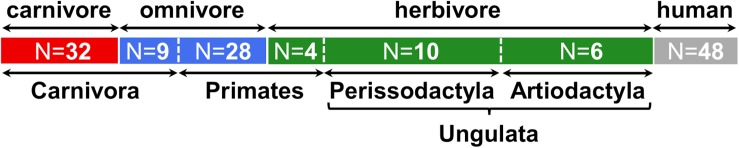
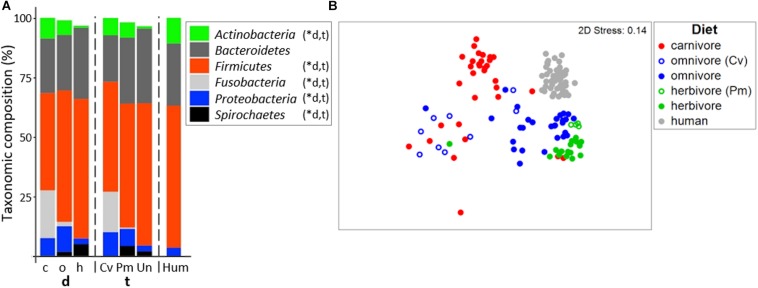
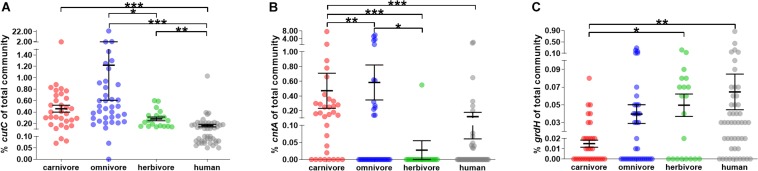
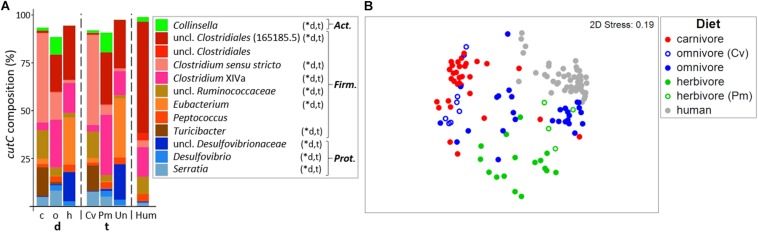
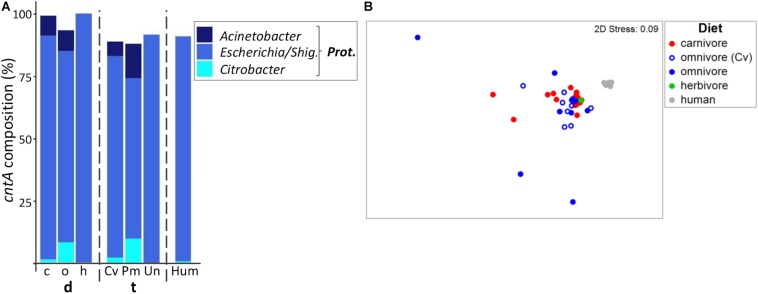
Human gut bacteria metabolize dietary components such as choline and carnitine to trimethylamine (TMA) that is subsequently oxidized to trimethylamine-N-oxide (TMAO) by hepatic enzymes. Increased plasma levels of TMAO are associated with the development of cardiovascular and renal disease. In this study, we applied gene-targeted assays in order to quantify (qPCR) and characterize (MiSeq) bacterial genes encoding enzymes responsible for TMA production, namely choline-TMA lyase (CutC), carnitine oxygenase (CntA) and betaine reductase (GrdH) in 89 fecal samples derived from various mammals spanning three dietary groups (carnivores, omnivores and herbivores) and four host orders (Carnivora, Primates, Artiodactyla and Perissodactyla). All samples contained potential TMA-producing bacteria, however, at low abundances (<1.2% of total community). The cutC gene was more abundant in omnivores and carnivores compared with herbivores. CntA was almost absent from herbivores and grdH showed lowest average abundance of all three genes. Bacteria harboring cutC and grdH displayed high diversities where sequence types affiliated with various taxa within Firmicutes dominated, whereas cntA comprised sequences primarily linked to Escherichia. Composition of TMA-forming communities was strongly influenced by diet and host taxonomy and despite their high correlation, both factors contributed uniquely to community structure. Furthermore, Random Forest (RF) models could differentiate between groups at high accuracies. This study gives a comprehensive overview of potential TMA-producing bacteria in the mammalian gut demonstrating that both diet and host taxonomy govern their abundance and composition. It highlights the role of functional redundancy sustaining potential TMA formation in distinct gut environments.
Keywords: betaine; carnitine; choline; diet; gut microbiota; mammals; microbial ecology; trimethylamine.
Copyright © 2020 Rath, Rud, Pieper and Vital.
Figures

References
-
- Clarke K. R., Gorley R. N., Somerfield P. J., Warwick R. M. (2014). Change in Marine Communities: An approach to Statistical Analysis and Interpretation, 3rd Edn Plymouth: PRIMER-E, 256.
-
- Costa M. C., Arroyo L. G., Allen-Vercoe E., Stämpfli H. R., Kim P. T., Sturgeon A., et al. (2012). Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3-V5 region of the 16s rRNA gene. PLoS One 7:e41484. 10.1371/journal.pone.0041484 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources