

Spatiotemporal control of phosphatidic acid signaling with optogenetic, engineered phospholipase Ds
- PMID: 31999306
- PMCID: PMC7054994
- DOI: 10.1083/jcb.201907013
Spatiotemporal control of phosphatidic acid signaling with optogenetic, engineered phospholipase Ds
Abstract
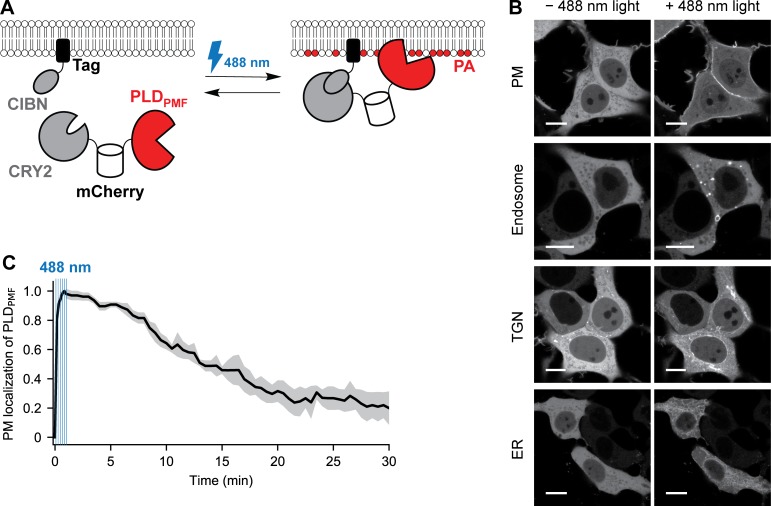
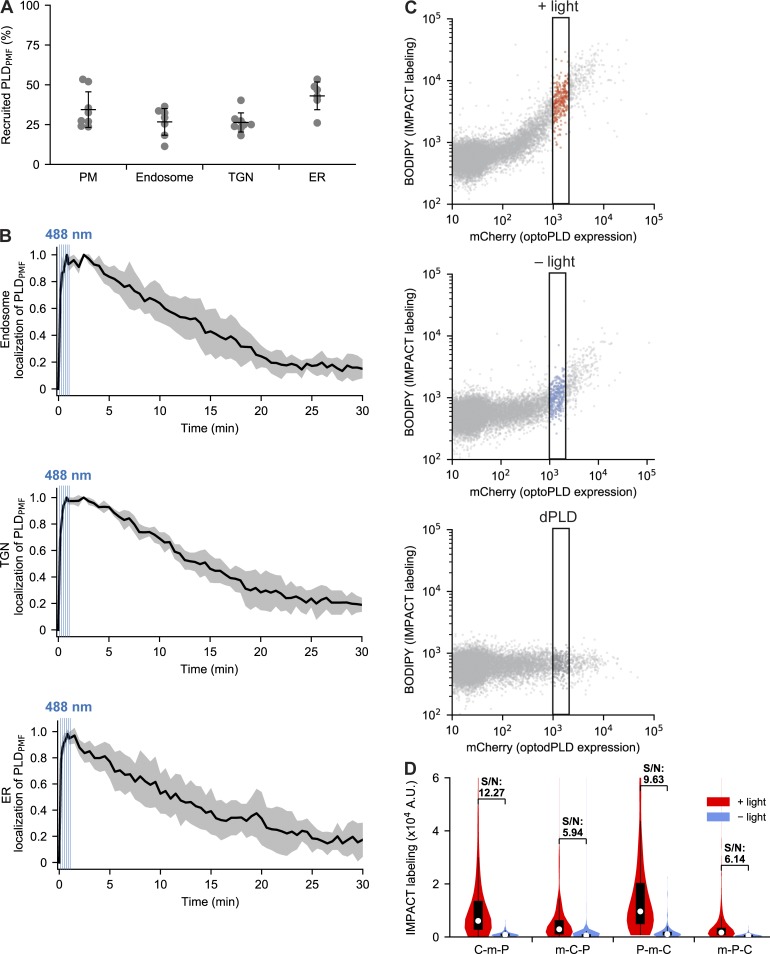
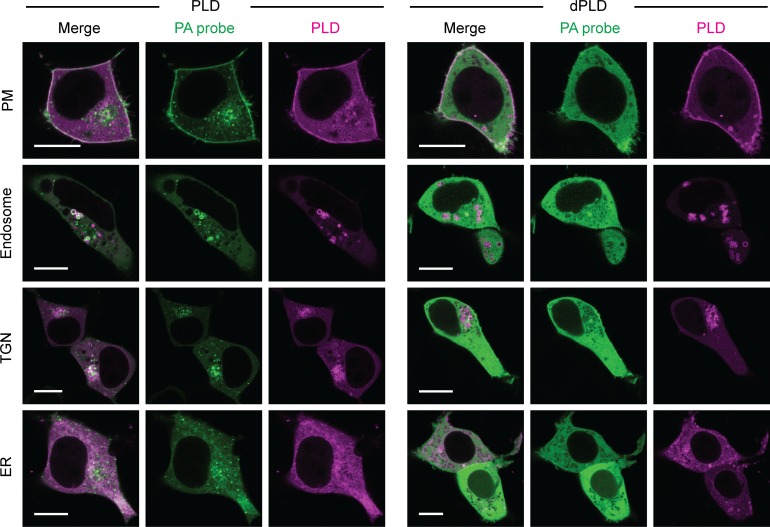
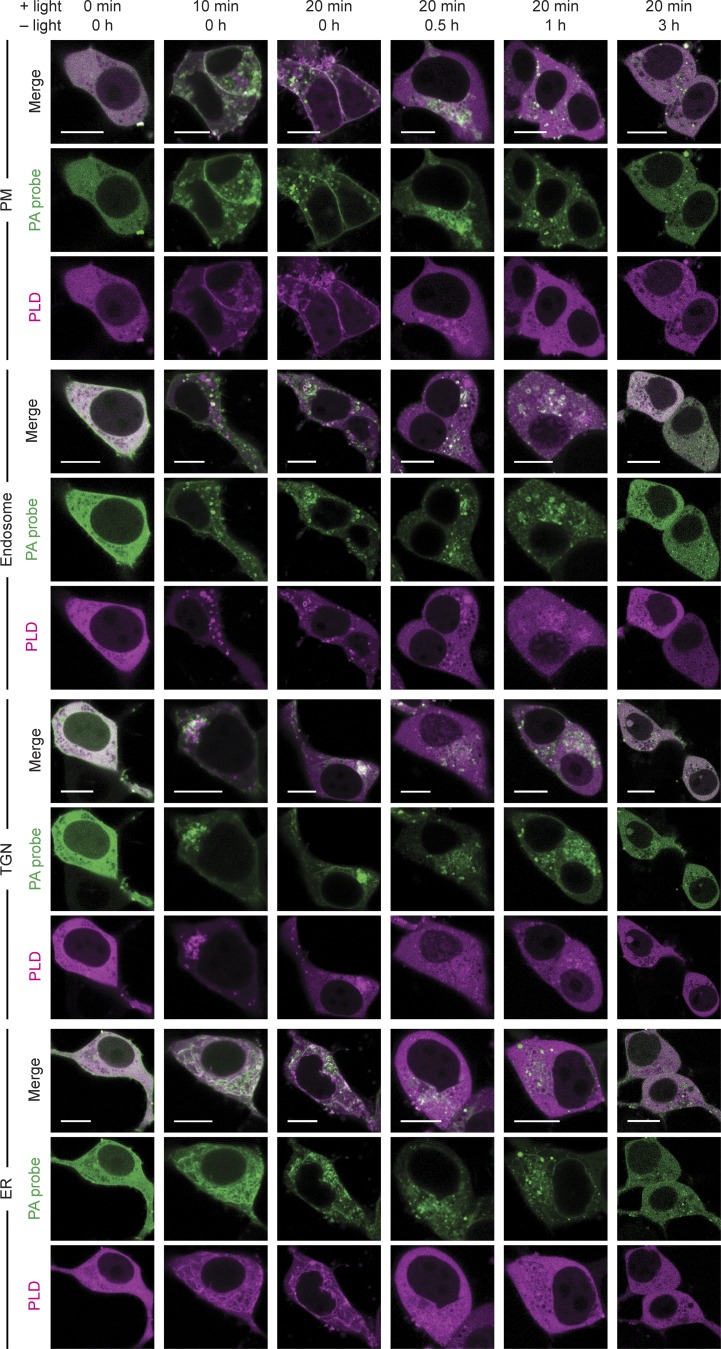
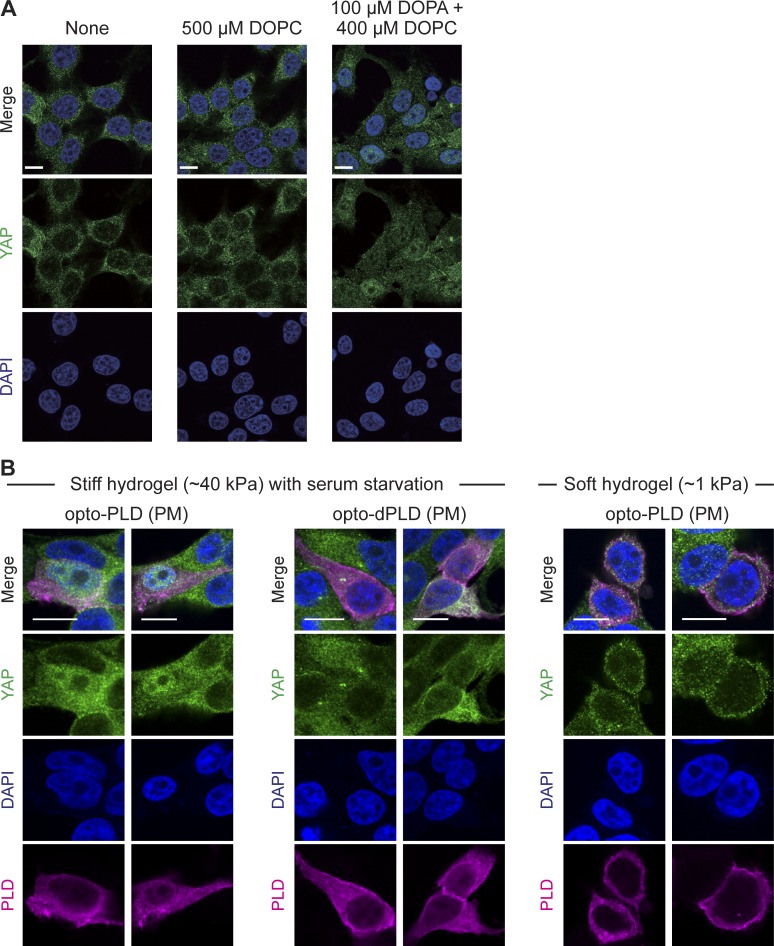
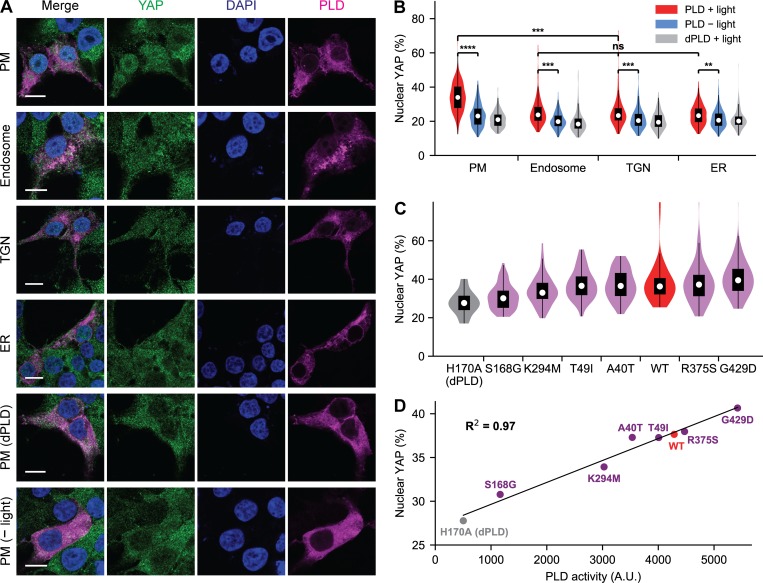
Phosphatidic acid (PA) is both a central phospholipid biosynthetic intermediate and a multifunctional lipid second messenger produced at several discrete subcellular locations. Organelle-specific PA pools are believed to play distinct physiological roles, but tools with high spatiotemporal control are lacking for unraveling these pleiotropic functions. Here, we present an approach to precisely generate PA on demand on specific organelle membranes. We exploited a microbial phospholipase D (PLD), which produces PA by phosphatidylcholine hydrolysis, and the CRY2-CIBN light-mediated heterodimerization system to create an optogenetic PLD (optoPLD). Directed evolution of PLD using yeast membrane display and IMPACT, a chemoenzymatic method for visualizing cellular PLD activity, yielded a panel of optoPLDs whose range of catalytic activities enables mimicry of endogenous, physiological PLD signaling. Finally, we applied optoPLD to elucidate that plasma membrane, but not intracellular, pools of PA can attenuate the oncogenic Hippo signaling pathway. OptoPLD represents a powerful and precise approach for revealing spatiotemporally defined physiological functions of PA.
© 2020 Tei and Baskin.
Figures

Similar articles
-
Imaging and Editing the Phospholipidome.Acc Chem Res. 2022 Nov 1;55(21):3088-3098. doi: 10.1021/acs.accounts.2c00510. Epub 2022 Oct 24. Acc Chem Res. 2022. PMID: 36278840 Free PMC article. Review.
-
Click chemistry and optogenetic approaches to visualize and manipulate phosphatidic acid signaling.J Biol Chem. 2022 Apr;298(4):101810. doi: 10.1016/j.jbc.2022.101810. Epub 2022 Mar 8. J Biol Chem. 2022. PMID: 35276134 Free PMC article.
-
A real-time, click chemistry imaging approach reveals stimulus-specific subcellular locations of phospholipase D activity.Proc Natl Acad Sci U S A. 2019 Jul 30;116(31):15453-15462. doi: 10.1073/pnas.1903949116. Epub 2019 Jul 16. Proc Natl Acad Sci U S A. 2019. PMID: 31311871 Free PMC article.
-
Regulation of the Hippo Pathway by Phosphatidic Acid-Mediated Lipid-Protein Interaction.Mol Cell. 2018 Oct 18;72(2):328-340.e8. doi: 10.1016/j.molcel.2018.08.038. Epub 2018 Oct 4. Mol Cell. 2018. PMID: 30293781 Free PMC article.
-
Phospholipase D/phosphatidic acid signal transduction: role and physiological significance in lung.Mol Cell Biochem. 2002 May-Jun;234-235(1-2):99-109. Mol Cell Biochem. 2002. PMID: 12162465 Review.
Cited by
-
Optical Control of Phosphatidic Acid Signaling.ACS Cent Sci. 2021 Jul 28;7(7):1205-1215. doi: 10.1021/acscentsci.1c00444. Epub 2021 Jul 14. ACS Cent Sci. 2021. PMID: 34345670 Free PMC article.
-
NME3 binds to phosphatidic acid and mediates PLD6-induced mitochondrial tethering.J Cell Biol. 2023 Oct 2;222(10):e202301091. doi: 10.1083/jcb.202301091. Epub 2023 Aug 16. J Cell Biol. 2023. PMID: 37584589 Free PMC article.
-
The dynamic regulatory network of phosphatidic acid metabolism: a spotlight on substrate cycling between phosphatidic acid and diacylglycerol.Biochem Soc Trans. 2024 Oct 30;52(5):2123-2132. doi: 10.1042/BST20231511. Biochem Soc Trans. 2024. PMID: 39417337 Free PMC article. Review.
-
Ultralow background membrane editors for spatiotemporal control of lipid metabolism and signaling.bioRxiv [Preprint]. 2023 Aug 31:2023.08.31.555787. doi: 10.1101/2023.08.31.555787. bioRxiv. 2023. Update in: ACS Cent Sci. 2024 Jan 30;10(3):543-554. doi: 10.1021/acscentsci.3c01105. PMID: 37693485 Free PMC article. Updated. Preprint.
-
Emerging Approaches for Studying Lipid Dynamics, Metabolism, and Interactions in Cells.Annu Rev Biochem. 2025 Jun;94(1):417-446. doi: 10.1146/annurev-biochem-083024-110827. Epub 2025 Mar 18. Annu Rev Biochem. 2025. PMID: 40101206 Review.
References
-
- Angelini A., Chen T.F., de Picciotto S., Yang N.J., Tzeng A., Santos M.S., Van Deventer J.A., Traxlmayr M.W., and Wittrup K.D.. 2015. Protein Engineering and Selection Using Yeast Surface Display. In Yeast Surface Display: Methods, Protocols, and Applications. B. Liu, editor. Springer, New York. 3–36. 10.1007/978-1-4939-2748-7_1 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
