

Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy
- PMID: 32000802
- PMCID: PMC6993488
- DOI: 10.1186/s12943-020-1144-6
Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy
Erratum in
-
Correction to: Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy.Mol Cancer. 2020 Feb 14;19(1):31. doi: 10.1186/s12943-020-01148-y. Mol Cancer. 2020. PMID: 32059674 Free PMC article.
Abstract
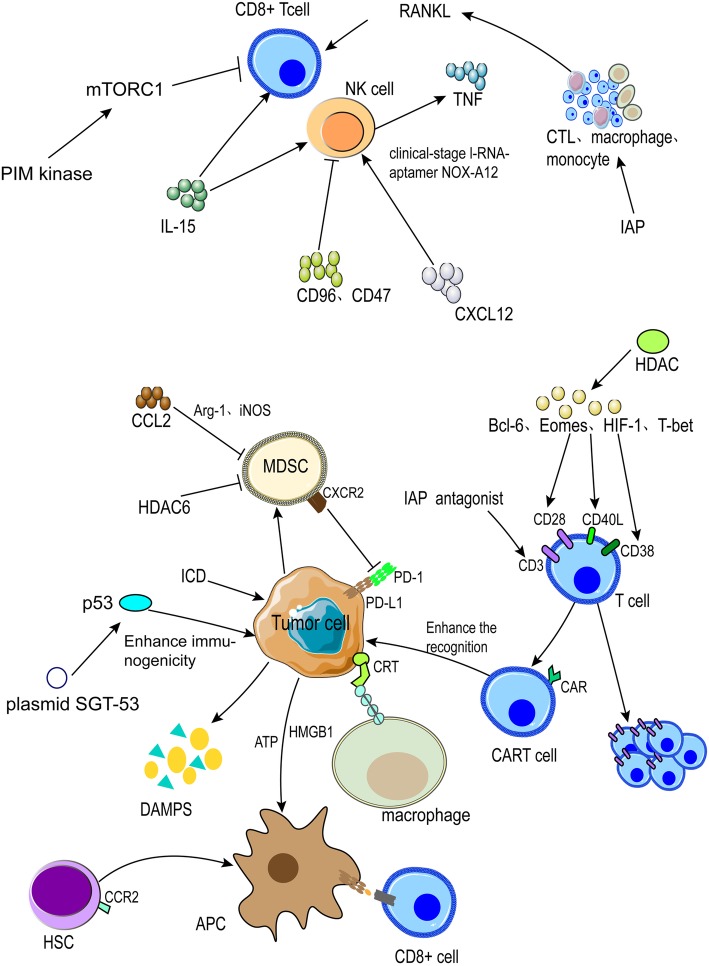
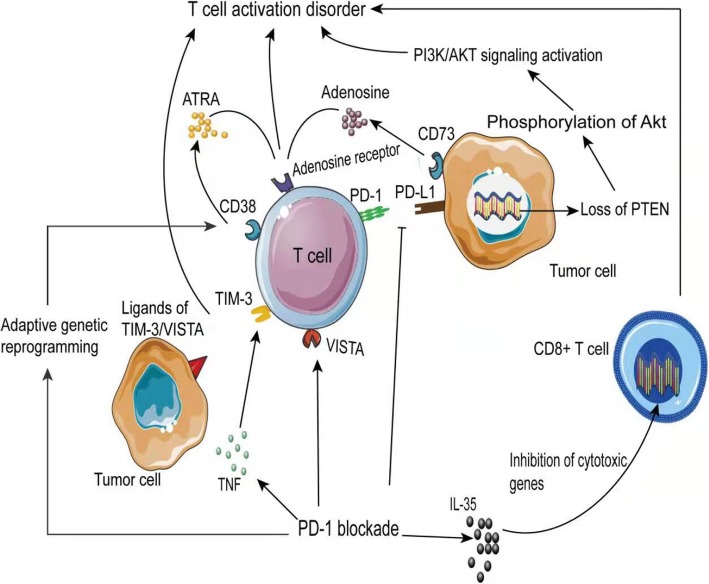
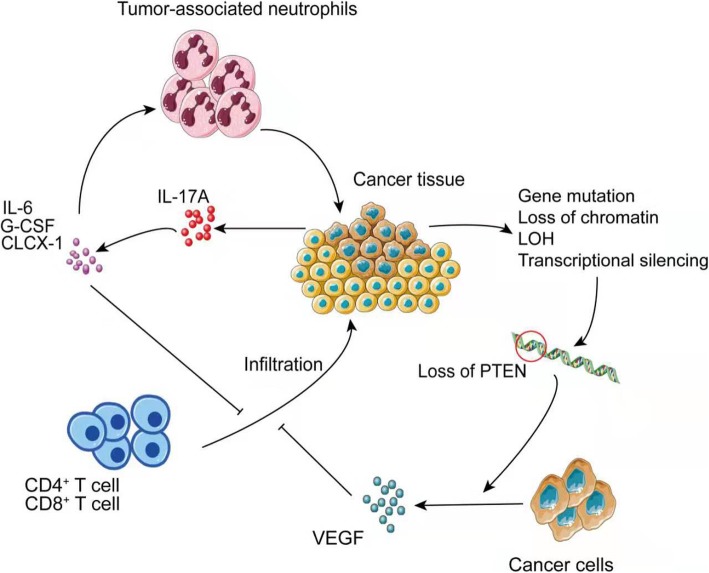
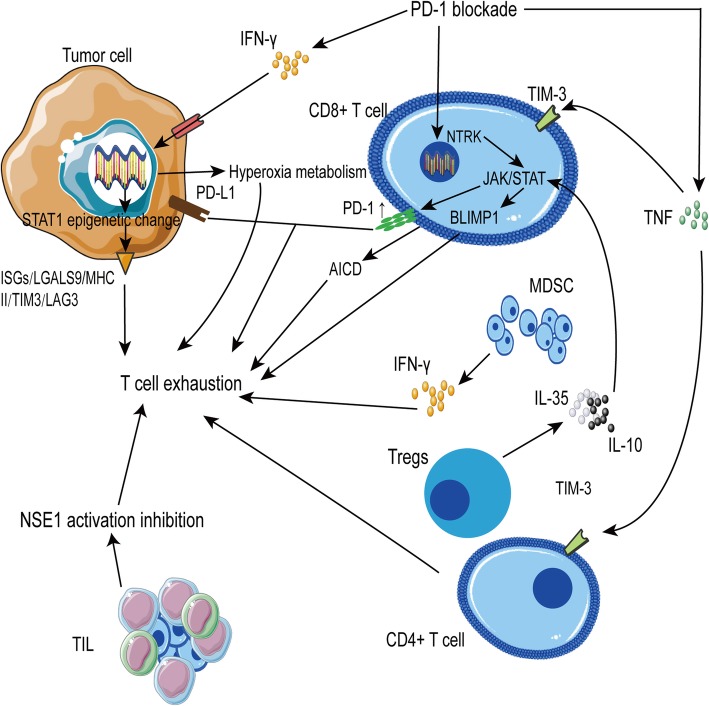
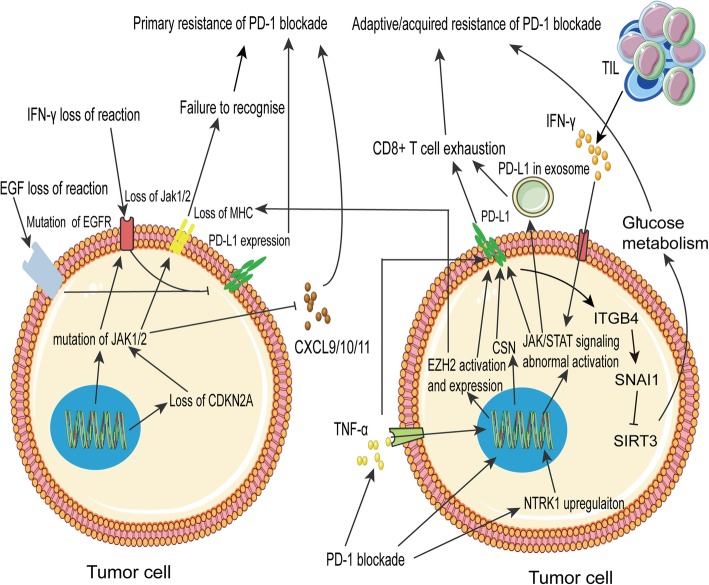
Immune checkpoint blockade targeting PD-1/PD-L1 has promising therapeutic efficacy in a variety of tumors, but resistance during treatment is a major issue. In this review, we describe the utility of PD-L1 expression levels, mutation burden, immune cell infiltration, and immune cell function for predicting the efficacy of PD-1/PD-L1 blockade therapy. Furthermore, we explore the mechanisms underlying immunotherapy resistance caused by PD-L1 expression on tumor cells, T cell dysfunction, and T cell exhaustion. Based on these mechanisms, we propose combination therapeutic strategies. We emphasize the importance of patient-specific treatment plans to reduce the economic burden and prolong the life of patients. The predictive indicators, resistance mechanisms, and combination therapies described in this review provide a basis for improved precision medicine.
Keywords: Cancer immunotherapy; Immune cells; Immune checkpoint blockade; PD-1/PD-L1; Precision medicine.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
-
- Jiang XJ, Wang J, Deng XY, Li XL, Li XY, Zeng ZY, et al. Immunotherapy targeted to immune checkpoint: a revolutionary breakthrough in cancer therapy. Prog Biochem Biophys. 2018;45:1178–1186.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
