

Wide spectrum and high frequency of genomic structural variation, including transposable elements, in large double-stranded DNA viruses
- PMID: 32002191
- PMCID: PMC6983493
- DOI: 10.1093/ve/vez060
Wide spectrum and high frequency of genomic structural variation, including transposable elements, in large double-stranded DNA viruses
Abstract
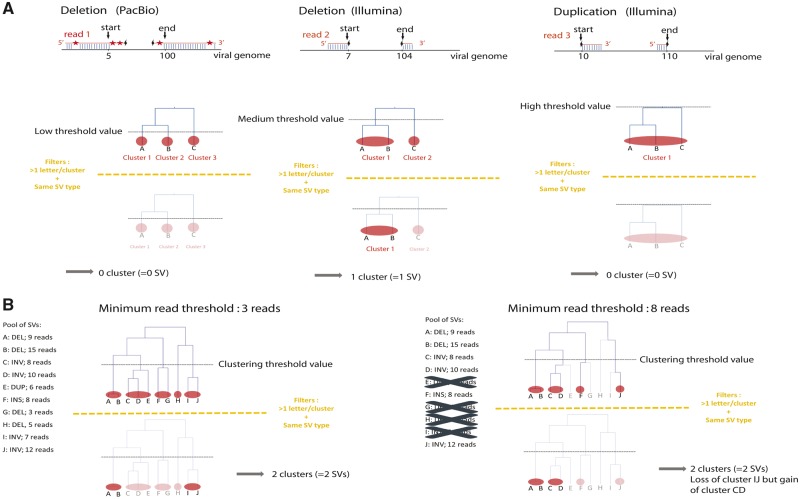
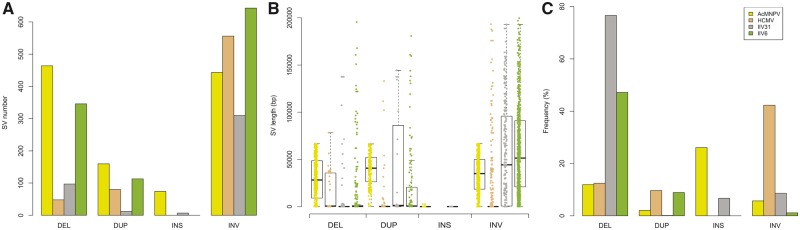
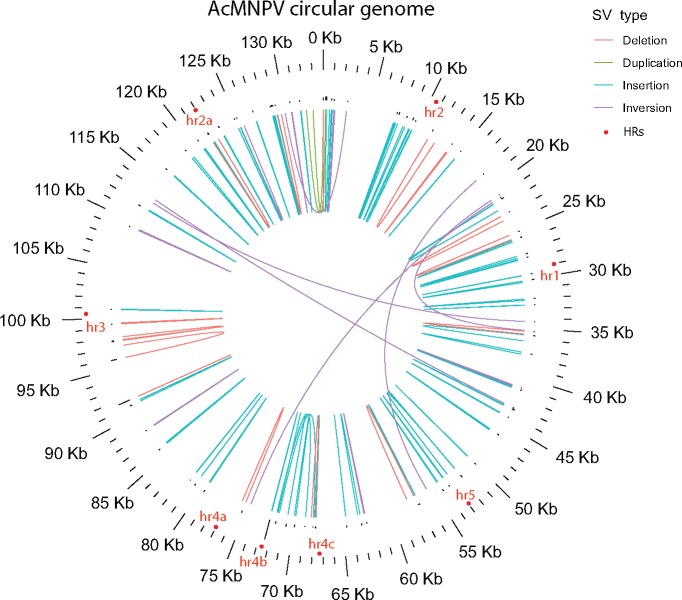
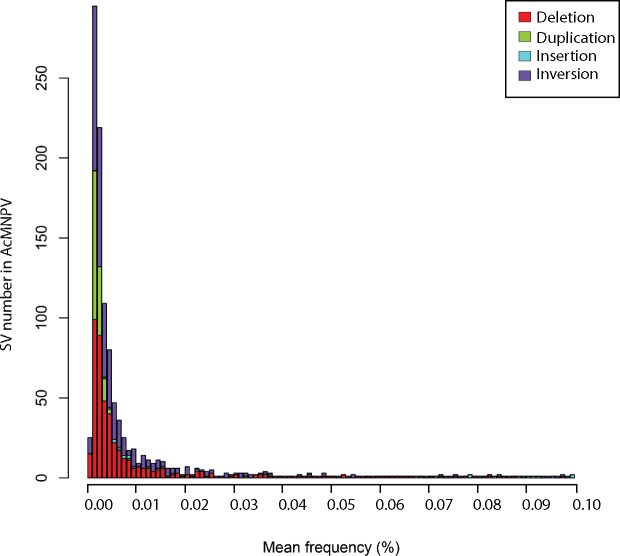
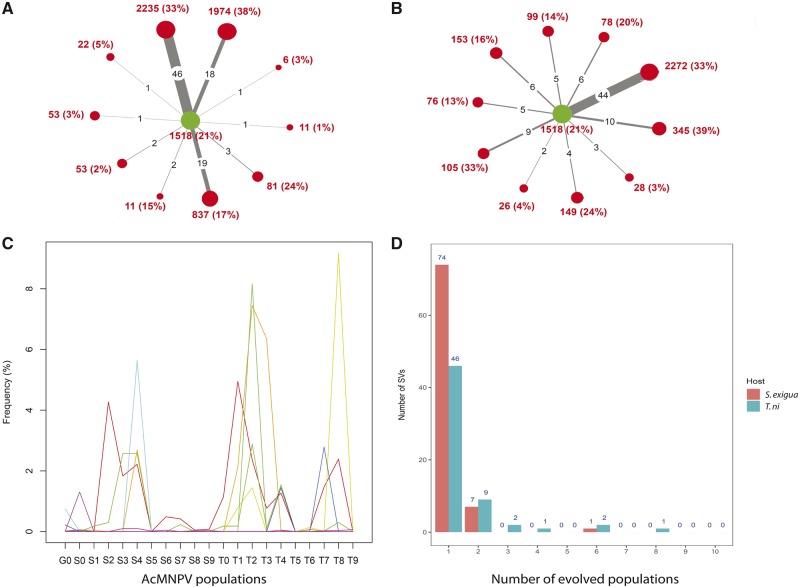
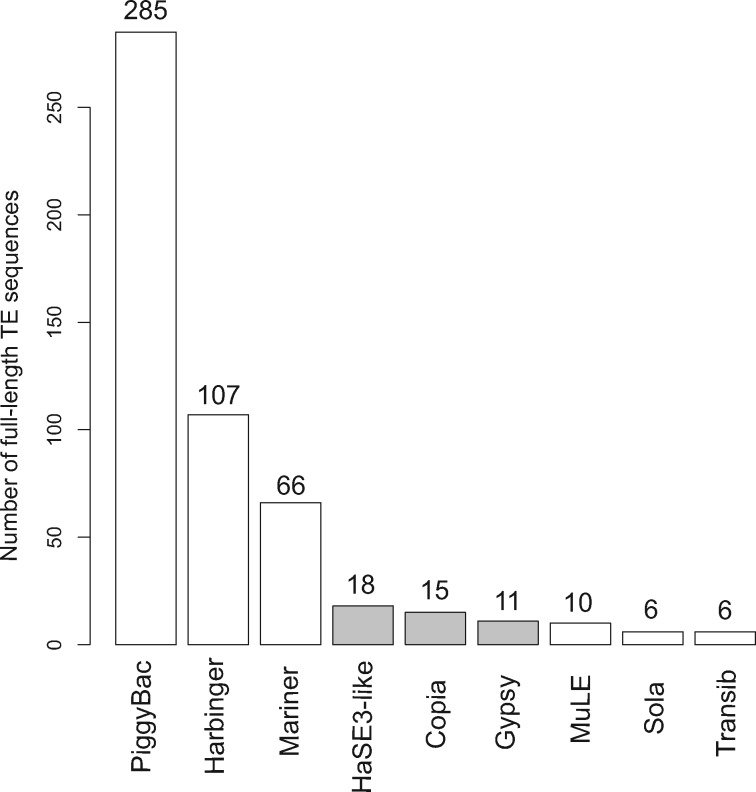
Our knowledge of the diversity and frequency of genomic structural variation segregating in populations of large double-stranded (ds) DNA viruses is limited. Here, we sequenced the genome of a baculovirus (Autographa californica multiple nucleopolyhedrovirus [AcMNPV]) purified from beet armyworm (Spodoptera exigua) larvae at depths >195,000× using both short- (Illumina) and long-read (PacBio) technologies. Using a pipeline relying on hierarchical clustering of structural variants (SVs) detected in individual short- and long-reads by six variant callers, we identified a total of 1,141 SVs in AcMNPV, including 464 deletions, 443 inversions, 160 duplications, and 74 insertions. These variants are considered robust and unlikely to result from technical artifacts because they were independently detected in at least three long reads as well as at least three short reads. SVs are distributed along the entire AcMNPV genome and may involve large genomic regions (30,496 bp on average). We show that no less than 39.9 per cent of genomes carry at least one SV in AcMNPV populations, that the vast majority of SVs (75%) segregate at very low frequency (<0.01%) and that very few SVs persist after ten replication cycles, consistent with a negative impact of most SVs on AcMNPV fitness. Using short-read sequencing datasets, we then show that populations of two iridoviruses and one herpesvirus are also full of SVs, as they contain between 426 and 1,102 SVs carried by 52.4-80.1 per cent of genomes. Finally, AcMNPV long reads allowed us to identify 1,757 transposable elements (TEs) insertions, 895 of which are truncated and occur at one extremity of the reads. This further supports the role of baculoviruses as possible vectors of horizontal transfer of TEs. Altogether, we found that SVs, which evolve mostly under rapid dynamics of gain and loss in viral populations, represent an important feature in the biology of large dsDNA viruses.
Keywords: baculovirus; genomic structural variation; herpesvirus; iridovirus; large double-stranded DNA viruses; transposable elements.
© The Author(s) 2020. Published by Oxford University Press.
Figures
Similar articles
-
Combined use of Oxford Nanopore and Illumina sequencing yields insights into soybean structural variation biology.BMC Biol. 2022 Feb 23;20(1):53. doi: 10.1186/s12915-022-01255-w. BMC Biol. 2022. PMID: 35197050 Free PMC article.
-
MetaSVs: A pipeline combining long and short reads for analysis and visualization of structural variants in metagenomes.Imeta. 2023 Oct 12;2(4):e139. doi: 10.1002/imt2.139. eCollection 2023 Nov. Imeta. 2023. PMID: 38868213 Free PMC article.
-
The Autographa californica Multiple Nucleopolyhedrovirus ac83 Gene Contains a cis-Acting Element That Is Essential for Nucleocapsid Assembly.J Virol. 2017 Feb 14;91(5):e02110-16. doi: 10.1128/JVI.02110-16. Print 2017 Mar 1. J Virol. 2017. PMID: 28031366 Free PMC article.
-
Geographic distribution and adaptive significance of genomic structural variants: an anthropological genetics perspective.Hum Biol. 2014 Fall;86(4):260-75. doi: 10.13110/humanbiology.86.4.0260. Hum Biol. 2014. PMID: 25959693 Review.
-
Challenges in studying genomic structural variant formation mechanisms: the short-read dilemma and beyond.Bioessays. 2011 Nov;33(11):840-50. doi: 10.1002/bies.201100075. Epub 2011 Sep 30. Bioessays. 2011. PMID: 21959584 Review.
Cited by
-
Transcription Profile and Genomic Variations of Oryctes Rhinoceros Nudivirus in Coconut Rhinoceros Beetles.J Virol. 2020 Oct 27;94(22):e01097-20. doi: 10.1128/JVI.01097-20. Print 2020 Oct 27. J Virol. 2020. PMID: 32878889 Free PMC article.
-
Spatially Segregated Transmission of Co-Occluded Baculoviruses Limits Virus-Virus Interactions Mediated by Cellular Coinfection during Primary Infection.Viruses. 2022 Jul 31;14(8):1697. doi: 10.3390/v14081697. Viruses. 2022. PMID: 36016318 Free PMC article.
-
Bacsnp: Using Single Nucleotide Polymorphism (SNP) Specificities and Frequencies to Identify Genotype Composition in Baculoviruses.Viruses. 2020 Jun 9;12(6):625. doi: 10.3390/v12060625. Viruses. 2020. PMID: 32526997 Free PMC article.
-
Generation of Variability in Chrysodeixis includens Nucleopolyhedrovirus (ChinNPV): The Role of a Single Variant.Viruses. 2021 Sep 22;13(10):1895. doi: 10.3390/v13101895. Viruses. 2021. PMID: 34696324 Free PMC article.
-
Monitoring Insect Transposable Elements in Large Double-Stranded DNA Viruses Reveals Host-to-Virus and Virus-to-Virus Transposition.Mol Biol Evol. 2021 Aug 23;38(9):3512-3530. doi: 10.1093/molbev/msab198. Mol Biol Evol. 2021. PMID: 34191026 Free PMC article.
References
-
- Ackermann H.-W., Smirnoff W. A. (1983) ‘A Morphological Investigation of 23 Baculoviruses’, Journal of Invertebrate Pathology, 41: 269–80.
Associated data
LinkOut - more resources
Full Text Sources
